
 PDF下载 ( 725 KB)
PDF下载 ( 725 KB)

β-葡萄糖醛酸酶(GUS)在原发性肝内胆管结石形成中的作用
DOI: 10.12449/JCH240530
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:李果、叶海军、李江负责课题设计,资料分析,撰写论文;徐至开、李文涛、徐承雷、李江参与收集数据,修改论文;李江负责拟定写作思路,指导撰写文章并最后定稿。
-
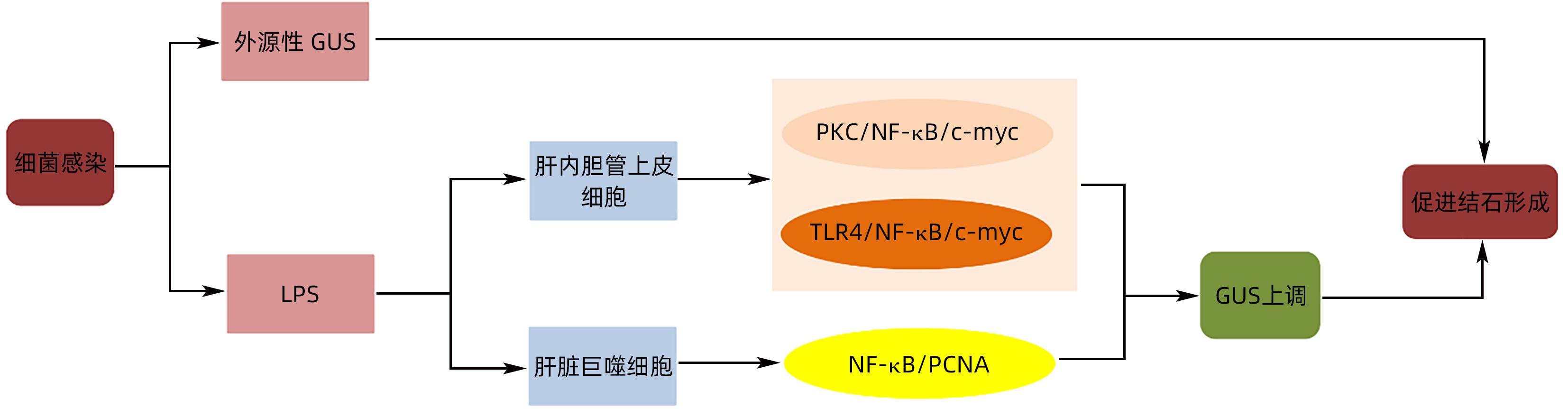
摘要: 原发性肝内胆管结石(PIS)是我国西南地区高发和难治性疾病,对患者的生活造成极大的影响。胆道系统慢性感染产生的β-葡萄糖醛酸酶等代谢产物,在色素性结石的形成中起着重要作用,除细菌产生的外源性β-葡萄糖醛酸酶,肝内胆管细胞产生的内源性β-葡萄糖醛酸酶在结石的形成中亦扮演重要角色。本文就PIS发病机制中β-葡萄糖醛酸酶的作用研究进展予以分析,以期为PIS的防治提供可能的方式。Abstract: Primary intrahepatic stones (PIS) is a refractory disease with a high incidence rate in Southwest China, which greatly affects the life of patients. Metabolites, such as β-glucuronidase produced by chronic biliary tract infection, play an important role in the formation of pigmented stones. In addition to exogenous β-glucuronidase produced by bacteria, endogenous β-glucuronidase produced by intrahepatic bile duct cells also plays an important role in the formation of stones. This article analyzes the research advances in the role of β-glucuronidase in the pathogenesis of PIS, in order to provide a possible method for the prevention and treatment of PIS.
-
Key words:
- Gallstones /
- Glucuronidase /
- Pathologic Processes
-
[1] HUANG ZQ, LIU YX. Surgical treatment of intrahepatic bile duct stones[J]. Chin J Pract Surg, 1997, 17( 3): 14- 18.黄志强, 刘永雄. 肝内胆管结石的外科治疗(40年回顾)[J]. 中国实用外科杂志, 1997, 17( 3): 14- 18. [2] TAZUMA S. Gallstone disease: Epidemiology, pathogenesis, and classification of biliary stones(common bile duct and intrahepatic)[J]. Best Pract Res Clin Gastroenterol, 2006, 20( 6): 1075- 1083. DOI: 10.1016/j.bpg.2006.05.009. [3] SUZUKI Y, MORI T, YOKOYAMA M, et al. Hepatolithiasis: Analysis of Japanese nationwide surveys over a period of 40 years[J]. J Hepato Biliary Pancreat, 2014, 21( 9): 617- 622. DOI: 10.1002/jhbp.116. [4] RAN X, YIN BB, MA BJ. Four major factors contributing to intrahepatic stones[J]. Gastroenterol Res Pract, 2017, 2017: 7213043. DOI: 10.1155/2017/7213043. [5] XIA H, ZHANG H, XIN X, et al. Surgical management of recurrence of primary intrahepatic bile duct stones[J]. Can J Gastroenterol Hepatol, 2023: 5158580. DOI: 10.1155/2023/5158580. [6] TANAKA Y, TAINAKA T, SUMIDA W, et al. The efficacy of resection of intrahepatic bile duct stenosis-causing membrane or septum for preventing hepatolithiasis after choledochal cyst excision[J]. J Pediatr Surg, 2017, 52( 12): 1930- 1933. DOI: 10.1016/j.jpedsurg.2017.08.056. [7] TSUI WMS, LAM PWY, LEE WK, et al. Primary hepatolithiasis, recurrent pyogenic cholangitis, and oriental cholangiohepatitis: A tale of 3 countries[J]. Adv Anat Pathol, 2011, 18( 4): 318- 328. DOI: 10.1097/PAP.0b013e318220fb75. [8] MOLINERO N, RUIZ L, MILANI C, et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile[J]. Microbiome, 2019, 7( 1): 100. DOI: 10.1186/s40168-019-0712-8. [9] REN JP, QIU JF, ZOU Y, et al. Research advances in the formation mechanism of primary intrahepatic stones caused by biliary flora[J]. J Clin Hepatol, 2022, 38( 2): 477- 482. DOI: 10.3969/j.issn.1001-5256.2022.02.045.任江平, 邱锦飞, 邹杨, 等. 胆道菌群导致原发性肝内胆管结石形成机理的研究进展[J]. 临床肝胆病杂志, 2022, 38( 2): 477- 482. DOI: 10.3969/j.issn.1001-5256.2022.02.045. [10] LIM JH. Liver flukes: The malady neglected[J]. Korean J Radiol, 2011, 12( 3): 269- 279. DOI: 10.3348/kjr.2011.12.3.269. [11] CHEN AN, WANG H, ZHOU YN, et al. Supervision of the main pathogenic bacteria resistance rates and rationality analysis of antibacterial drugs usage in hepatolithiasis with biliary tract infection[J]. J Guangdong Pharm Univ, 2017, 33( 3): 388- 392. DOI: 10.16809/j.cnki.2096-3653.2017011204.陈安妮, 王慧, 周燕妮, 等. 肝内胆管结石病原菌监测及抗菌药物使用合理性分析[J]. 广东药学院学报, 2017, 33( 3): 388- 392. DOI: 10.16809/j.cnki.2096-3653.2017011204. [12] WU ZQ. Bacterial spectrum of bile in hepatolithiasis patients and it’s relationship with the infection after operation[D]. Fuzhou: Fujian Medical University, 2017.吴樟强. 肝内胆管结石患者胆汁细菌谱与术后感染的关系[D]. 福州: 福建医科大学, 2017. [13] CLEMENTE G, de ROSE AM, MURRI R, et al. Liver resection for primary intrahepatic stones: Focus on postoperative infectious complications[J]. World J Surg, 2016, 40( 2): 433- 439. DOI: 10.1007/s00268-015-3227-x. [14] MAKI T. Pathogenesis of calcium bilirubinate gallstone: Role of E. coli, beta-glucuronidase and coagulation by inorganic ions, polyelectrolytes and agitation[J]. Ann Surg, 1966, 164( 1): 90- 100. DOI: 10.1097/00000658-196607000-00010. [15] HUANG ZQ, YANG KZ, MENG XJ, et al. The significance of bile β- glucuronidase activity[J]. Chin J Surg, 1982, 20( 1): 49- 52, 64.黄志强, 杨可桢, 孟宪钧, 等. 胆汁β-葡萄糖醛酸酶活性的意义[J]. 中华外科杂志, 1982, 20( 1): 49- 52, 64. [16] WANG PP, JIA YF, WU RR, et al. Human gut bacterial β-glucuronidase inhibition: An emerging approach to manage medication therapy[J]. Biochem Pharmacol, 2021, 190: 114566. DOI: 10.1016/j.bcp.2021.114566. [17] CHEN ZQ, TANG S, ZHANG CX, et al. Research progress on the interaction between gut bacterial β-glucuronidase and Chinese herbal medicine[J]. Acta Pharm Sin B, 2022, 57( 12): 3465- 3479. DOI: 10.16438/j.0513-4870.2022-1091.陈智强, 汤帅, 张畅煊, 等. 肠道菌群β-葡萄糖醛酸苷酶与中草药的互作关系研究进展[J]. 药学学报, 2022, 57( 12): 3465- 3479. DOI: 10.16438/j.0513-4870.2022-1091. [18] FEVERY J, BLANCKAERT N, LEROY P, et al. Analysis of bilirubins in biological fluids by extraction and thin-layer chromatography of the intact tetrapyrroles: Application to bile of patients with gilbert’s syndrome, hemolysis, or cholelithiasis[J]. Hepatology, 2007, 3( 2): 177- 183. DOI: 10.1002/hep.1840030207. [19] VÍTEK L, OSTROW JD. Bilirubin chemistry and metabolism; harmful and protective aspects[J]. Curr Pharm Des, 2009, 15( 25): 2869- 2883. DOI: 10.2174/138161209789058237. [20] TANG W, LU HY, SUN Q, et al. Characteristics of gut microbiota and its association with the activity of β-glucuronidase in neonates with hyperbilirubinemia[J]. Chin J Contemp Pediatr, 2021, 23( 7): 677- 683. DOI: 10.7499/j.issn.1008-8830.2102039.唐炜, 卢红艳, 孙勤, 等. 高胆红素血症新生儿肠道菌群特点及与β-葡萄糖醛酸苷酶活性的相关性[J]. 中国当代儿科杂志, 2021, 23( 7): 677- 683. DOI: 10.7499/j.issn.1008-8830.2102039. [21] NIE YF, HU J, YAN XH. Cross-talk between bile acids and intestinal microbiota in host metabolism and health[J]. J Zhejiang Univ Sci B, 2015, 16( 6): 436- 446. DOI: 10.1631/jzus.B1400327. [22] ROUGIER P, BUGAT R, DOUILLARD JY, et al. Phase II study of irinotecan in the treatment of advanced colorectal cancer in chemotherapy-naive patients and patients pretreated with fluorouracil-based chemotherapy[J]. J Clin Oncol, 1997, 15( 1): 251- 260. DOI: 10.1200/jco.1997.15.1.251. [23] BHATT AP, PELLOCK SJ, BIERNAT KA, et al. Targeted inhibition of gut bacterial β-glucuronidase activity enhances anticancer drug efficacy[J]. Proc Natl Acad Sci USA, 2020, 117( 13): 7374- 7381. DOI: 10.1073/pnas.1918095117. [24] SAITTA KS, ZHANG C, LEE KK, et al. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: Mode of action and pharmacokinetics[J]. Xenobiotica, 2014, 44( 1): 28- 35. DOI: 10.3109/00498254.2013.811314. [25] ELMASSRY MM, KIM S, BUSBY B. Predicting drug-metagenome interactions: Variation in the microbial β-glucuronidase level in the human gut metagenomes[J]. PLoS One, 2021, 16( 1): e0244876. DOI: 10.1371/journal.pone.0244876. [26] CHEN BR, FU SW, LU LG, et al. A preliminary study of biliary microbiota in patients with bile duct stones or distal cholangiocarcinoma[J]. Biomed Res Int, 2019, 2019: 1092563. DOI: 10.1155/2019/1092563. [27] SHEN HZ, YE FQ, XIE L, et al. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria[J]. Sci Rep, 2015, 5: 17450. DOI: 10.1038/srep17450. [28] STRINGER AM, GIBSON RJ, LOGAN RM, et al. Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats[J]. Cancer Biol Ther, 2008, 7( 12): 1919- 1925. DOI: 10.4161/cbt.7.12.6940. [29] CHEN KW, YUAN TM. The role of microbiota in neonatal hyperbilirubinemia[J]. Am J Transl Res, 2020, 12( 11): 7459- 7474. [30] LI Y, SONG JM, YU H, et al. PGE(2) induces MUC2 and MUC5AC expression in human intrahepatic biliary epithelial cells via EP4/p38MAPK activation[J]. Ann Hepatol, 2013, 12( 3): 479- 486. [31] LI FY, CHENG NS, CHENG JQ, et al. Proliferating cell nuclear antigen shRNA treatment attenuates chronic proliferative cholangitis in rats[J]. J Gastro And Hepatol, 2009, 24( 5): 920- 926. DOI: 10.1111/j.1440-1746.2008.05688.x. [32] OYABU H, TABATA M, NAKAYAMA F. Nonbacterial transformation of bilirubin in bile[J]. Dig Dis Sci, 1987, 32( 8): 809- 816. DOI: 10.1007/BF01296701. [33] HO KJ, HSU SC, CHEN JS, et al. Human biliary β-glucuronidase: Correlation of its activity with deconjugation of bilirubin in the bile[J]. Eur J Clin Investigation, 1986, 16( 5): 361- 367. DOI: 10.1111/j.1365-2362.1986.tb01010.x. [34] WAUTERS L, CEULEMANS M, LAMBAERTS M, et al. Association between duodenal bile salts and gastric emptying in patients with functional dyspepsia[J]. Gut, 2021, 70( 11): 2208- 2210. DOI: 10.1136/gutjnl-2020-323598. [35] ZHANG XB, CUI NQ, LI DH. Effect of clearing heat and removing dampness method on formation of pigment gallstones in rabbits[J]. Chin J Integr Trad West Med, 2007, 27( 3): 241- 243. DOI: 10.3321/j.issn:1003-5370.2007.03.014.张西波, 崔乃强, 李东华. 清热利湿方防治兔胆色素结石形成的实验研究[J]. 中国中西医结合杂志, 2007, 27( 3): 241- 243. DOI: 10.3321/j.issn:1003-5370.2007.03.014. [36] YAO DB, DONG QZ, TIAN Y, et al. Lipopolysaccharide stimulates endogenous β-glucuronidase via PKC/NF-κB/c-myc signaling cascade: A possible factor in hepatolithiasis formation[J]. Mol Cell Biochem, 2018, 444( 1): 93- 102. DOI: 10.1007/s11010-017-3234-3. [37] YAO CH. The role of LINC00311 in LPS-induced human intrahepatic bile duct epithelial cells to activate TLR4/NF-κB/c-myc signaling pathway to up-regulate the expression of endogenous β-glucuronidase[D]. Shenyang: China Medical University, 2019.姚晨辉. LINC00311在LPS诱导人肝内胆管上皮细胞激活TLR4/NF-κB/c-myc信号通路从而上调内源性β-葡萄糖醛酸酶表达过程中的作用[D]. 沈阳: 中国医科大学, 2019. [38] DONG XT, LIU JQ, XU YP, et al. Role of macrophages in experimental liver injury and repair in mice(Review)[J]. Exp Ther Med, 2019: 3835- 3847. DOI: 10.3892/etm.2019.7450. [39] TACKE F. Targeting hepatic macrophages to treat liver diseases[J]. J Hepatol, 2017, 66( 6): 1300- 1312. DOI: 10.1016/j.jhep.2017.02.026. [40] JIN CG, JIANG FR, ZHANG J, et al. Role of osteopontin in diet-induced brown gallstone formation in rats[J]. Chin Med J, 2021, 134( 9): 1093- 1100. DOI: 10.1097/CM9.0000000000001519. [41] YANG Y, WANG Y, WANG CF, et al. Macrophages and derived-TNF-α promote lipopolysaccharide-induced upregulation of endogenous β-glucuronidase in the epithelial cells of the bile duct: A possible facilitator of hepatolithiasis formation[J]. Clin Res Hepatol Gastroenterol, 2023, 47( 1): 102062. DOI: 10.1016/j.clinre.2022.102062. -

 下载:
下载:
 本文二维码
本文二维码
图(1)
计量
- 文章访问数: 1583
- HTML全文浏览量: 344
- PDF下载量: 119
- 被引次数: 0

