
 PDF下载 ( 769 KB)
PDF下载 ( 769 KB)

肝豆状核变性不同基因型患者的肝病表型及临床特征分析
DOI: 10.12449/JCH240819
Liver disease phenotypes and clinical features of patients with different genotypes of Wilson’s disease
-
摘要:
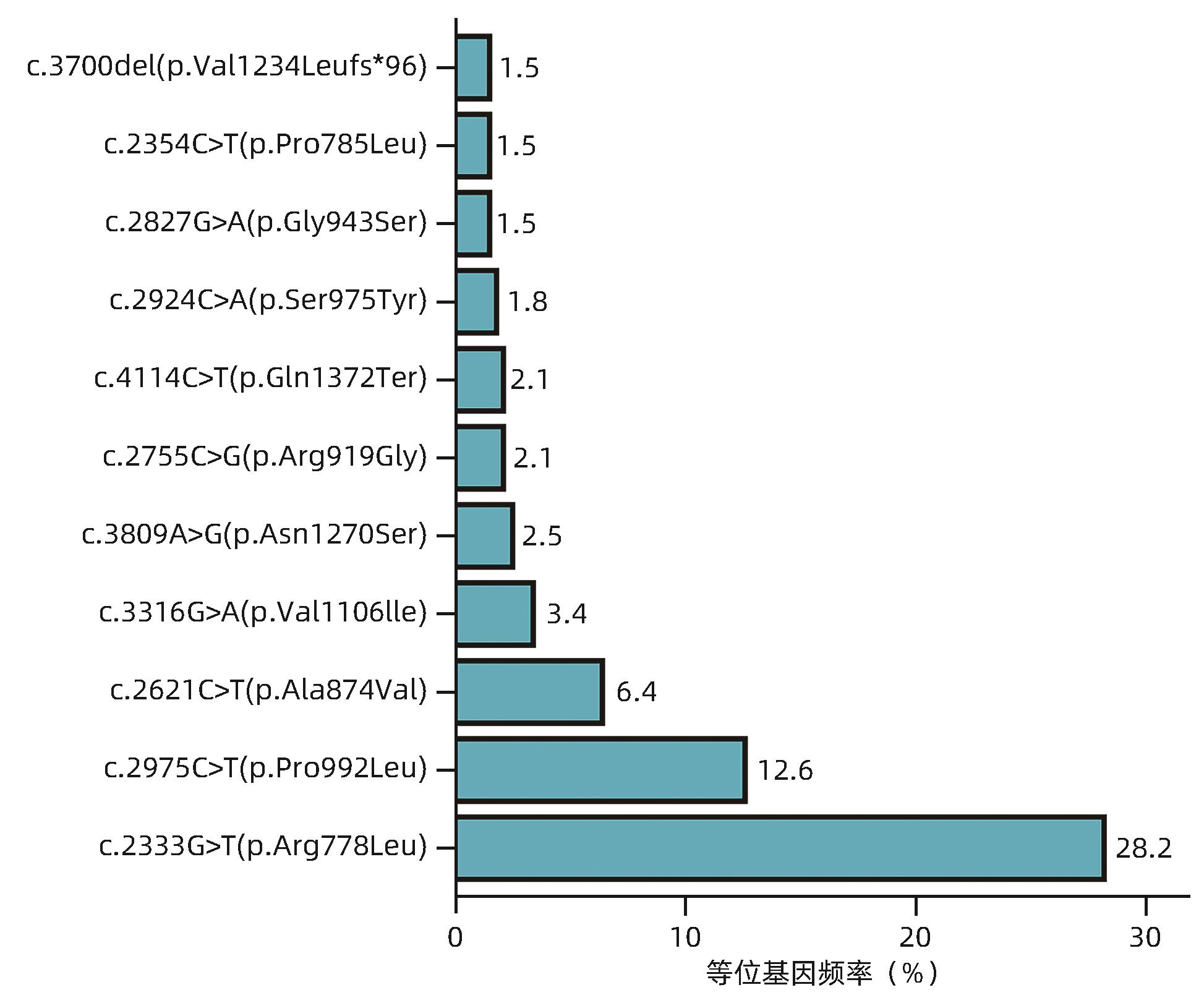
目的 研究肝豆状核变性(WD)不同基因型患者的肝病表型及临床特征。 方法 选取2008年8月—2023年6月在解放军总医院第五医学中心确诊并进行基因检测的163例WD患者,收集临床表现、实验室检查、病理学检查、影像学检查和ATP7B基因检测结果。根据ATP7B基因突变情况将患者分为R778L突变组和非R778L突变组;P992L突变组和非P992L突变组;截断突变组和非截断突变组。分析ATP7B基因c.2333G>T/p.R778L突变(R778L突变)、c.2975C>T/p.P992L突变(P992L突变)以及截断突变患者的肝病表型和临床特征。计量资料组间比较采用Mann-Whitney U检验或Kruskal-Wallis H检验。计数资料组间比较采用χ2检验或Fisher确切概率法。 结果 163例WD患者均表现为不同严重程度的肝病表型,121例(74.23%)被临床诊断为慢性肝病,36例(22.09%)为失代偿期肝硬化,6例(3.68%)为暴发性WD;此外,有5例(2例慢性肝病,3例失代偿期肝硬化)合并神经系统异常。163例WD患者最常见的ATP7B基因突变为R778L突变(等位基因频率为28.2%),其次为P992L突变(等位基因频率为12.6%),截断突变的等位基因频率为11.0%。3种突变在不同肝病表型之间的分布差异均无统计学意义(P值均>0.05)。R778L突变组的铜蓝蛋白水平显著低于非R778L突变组[0.04(0.02~0.08)g/L vs 0.08(0.03~0.13)g/L,Z=-2.889,P=0.004]。P992L组的ALT[135.0(80.5~237.0)U/L vs 80.5(36.0~173.3) U/L,Z=2.684,P=0.007]和AST[121.4(77.0~195.0)U/L vs 84.0(39.0~123.3)U/L,Z=3.388,P<0.001]均显著高于非P992L突变组。截断突变组的铜蓝蛋白[0.03(0.02~0.08)g/L vs 0.06(0.03~0.11)g/L,Z=-3.136,P=0.002]和血清铜[3.20(2.15~5.00)mg/L vs 4.20(2.60~7.50)mg/L,Z=-2.296,P=0.025]水平均显著低于非截断突变组。 结论 R778L突变、P992L突变和截断突变均与WD患者的肝病表型无关;但R778L突变与较低的铜蓝蛋白水平相关,P992L突变与较高的ALT和AST水平相关,截断突变与较低的铜蓝蛋白和血清铜水平相关。 Abstract:Objective To investigate the liver disease phenotypes and clinical features of patients with different genotypes of Wilson’s disease (WD). Methods A retrospective analysis was performed for 163 patients with WD who were diagnosed and underwent genetic testing in The Fifth Medical Center of Chinese PLA General Hospital from August 2008 to June 2023, and clinical manifestations, laboratory examination, pathological examination, imaging examination, and ATP7B genetic testing results were collected. According to ATP7B gene mutation, the patients were divided into groups as follows: R778L mutation group and non-R778L mutation group; P992L mutation group and non-P992L mutation group; truncation mutation group and non-truncation mutation group. Liver disease phenotypes and clinical features were analyzed for the patients with c.2333G>T/p.R778L mutation (R778L mutation), c.2975C>T/p.P992L mutation (P992L mutation), and truncation mutation of the ATP7B gene. The Mann-Whitney U test or the Kruskal-Wallis H test was used for comparison of continuous data between groups, and the chi-square test or the Fisher’s exact test was used for comparison of categorical data between groups. Results The 163 patients with WD had varying severities of liver disease phenotypes, among whom 121 (74.23%) were diagnosed with chronic liver disease, 36 (22.09%) were diagnosed with decompensated cirrhosis, and 6 (3.68%) were diagnosed with fulminant WD, and in addition, there were 5 patients (2 with chronic liver disease and 3 with decompensated cirrhosis) with neurological abnormalities. For the 163 patients with WD, R778L mutation (with an allele frequency of 28.2%) was the most common mutation in the ATP7B gene, followed by P992L mutation (with an allele frequency of 12.6%), and truncation mutation showed an allele frequency of 11.0%. There was no significant difference in the distribution of the three mutations across different liver disease phenotypes (P>0.05). The R778L mutation group had a significantly lower level of ceruloplasmin (CP) than the non-R778L mutation group [0.04 (0.02 — 0.08) g/L vs 0.08 (0.03 — 0.13) g/L, Z=-2.889, P=0.004]. Compared with the non-P992L mutation group, the P992L mutation group had significantly higher levels of alanine aminotransferase [135.0 (80.5 — 237.0) U/L vs 80.5 (36.0 — 173.3) U/L, Z=2.684, P=0.007] and aspartate aminotransferase [121.4 (77.0 — 195.0) U/L vs 84.0 (39.0 — 123.3) U/L, Z=3.388, P<0.001]. Compared with the non-truncation mutation group, the truncation mutation group had significantly lower levels of CP [0.03 (0.02 — 0.08) g/L vs 0.06 (0.03 — 0.11) g/L, Z=-3.136, P=0.002] and serum copper [3.20 (2.15 — 5.00) mg/L vs 4.20 (2.60 — 7.50) mg/L, Z=-2.296, P=0.025]. Conclusion R778L mutation, P992L mutation and truncation mutation are not associated with liver disease phenotype in WD patients; however, R778L mutation is associated with a lower level of CP, P992L mutation is associated with higher levels of ALT and AST, and truncation mutation is associated with lower levels of CP and serum copper. -
Key words:
- Hepatolenticular Degeneration /
- Genotype /
- Phenotype
-
表 1 不同肝病表型WD患者的临床特征比较
Table 1. Comparison of clinical features of WD patients with different liver disease phenotypes
项目 慢性肝病
(n=121)
失代偿期肝硬化
(n=36)
暴发性WD
(n=6)
统计值 P值 男[例(%)] 78(64.5) 13(36.1) 3(50.0) χ2=9.239 0.008 发病年龄(岁) 6.0(4.0~11.0) 13.5(12.0~34.0) 12.0(9.5~15.2) H=41.830 <0.001 体征[例(%)] 乏力、纳差 10(8.26) 27(75.00) 5(83.33) χ2=70.043 <0.001 腹痛、腹胀 12(9.92) 20(55.56) 4(66.67) χ2=36.878 <0.001 皮肤、巩膜黄染 3(2.48) 22(61.11) 3(50.00) χ2=64.447 <0.001 双下肢水肿 2(1.65) 14(38.89) 2(33.33) χ2=37.213 <0.001 K-F环阳性[例(%)]1) 22(19.6) 31(88.6) 6(100.0) χ2=66.403 <0.001 神经系统异常(例) χ2=3.989 0.170 四肢震颤 2 3 0 言语不清、语速缓慢 0 2 0 动作迟缓、走姿异常 0 2 0 实验室检查 ALT(U/L) 134.0(64.0~233.0) 36.0(26.8~55.0) 103.0(53.2~128.0) H=33.515 <0.001 AST(U/L) 97.0(55.0~140.0) 66.0(38.8~114.0) 176.0(97.5~248.0) H=7.192 0.026 TBil(μmol/L) 8.5(6.1~11.8) 29.4(18.4~50.3) 143.0(86.0~180.0) H=74.436 <0.001 Alb(g/L) 40.0(37.0~42.0) 29.0(26.0~34.2) 24.5(22.5~25.0) H=52.276 <0.001 WBC(×109/L) 5.94(5.29~7.05) 3.70(2.65~4.62) 7.56(5.66~8.75) H=35.293 <0.001 INR 1.02(0.96~1.09) 1.52(1.32~1.83) 1.96(1.83~2.07) H=72.887 <0.001 铜生化 24 h尿铜(μg/d)2) 138(88~244) 404(173~956) 544(507~629) H=20.913 <0.001 铜蓝蛋白(g/L) 0.04(0.02~0.11) 0.08(0.04~0.12) 0.10(0.08~0.11) H=5.331 0.070 血清铜(mg/L) 3.80(2.30~6.05) 5.30(3.42~7.12) 8.00(7.12~10.20) H=7.121 0.026 BMI(kg/m2) 16.5(14.9~20.0) 19.4(17.6~23.7) 24.1(20.0~25.0) H=43.325 <0.001 LSM(kPa)3) 6.5(4.9~11.1) 20.9(17.5~24.8) 24.8(20.7~26.2) H=16.973 <0.001 注:1)153例患者进行了眼科裂隙灯检查,其中慢性肝病组112例,失代偿期肝硬化组35例,暴发性WD组6例;2)141例患者进行了24 h尿铜水平检测,其中慢性肝病组107例,失代偿期肝硬化组30例,暴发性WD组4例;3)107例患者进行了肝脏弹性检查,慢性肝病组82例,失代偿期肝硬化组22例,暴发性WD组3例。
 下载: 导出CSV
下载: 导出CSV
表 2 163例WD患者的ATP7B基因突变情况
Table 2. Mutation of ATP7B gene in 163 patients with WD
突变 例数 纯合子 R778L/R778L 14 P992L/P992L 2 V890M/V890M 1 S975Y/S975Y 1 V606G/V606G 1 Q1372Ter/Q1372Ter 1 c.1708-5T>G/c.1708-5T>G 1 复合杂合子 R778L/P992L 13 R778L/V1106I 6 R778L/A874V 3 R778L/N1270S 3 R778L/Q1372Ter 3 R778L/其他突变1) 33 其他突变/其他突变 76 杂合子 R778L/第二突变未知 3 P785L/第二突变未知 1 c.1870-2A>G/第二突变未知 1 注:1)R778L与其他突变复合杂合子各自的例数均<3。
下载: 导出CSV
表 3 R778L突变、P992L突变、截断突变的WD患者肝病表型分析
Table 3. Phenotype analysis of liver disease in WD patients with R778L mutation, P992L mutation and truncation mutation
突变 慢性肝病(n=121) 失代偿期肝硬化(n=36) 暴发性WD(n=6) P值 R778L突变[例(%)] 0.953 是 57(47.11) 18(50.00) 3(50.00) 否 64(52.89) 18(50.00) 3(50.00) P992L突变[例(%)] 0.238 是 33(27.27) 5(13.89) 1(16.67) 否 88(72.73) 31(86.11) 5(83.33) 截断突变[例(%)] 0.453 是 23(19.01) 9(25.00) 0(0.00) 否 98(80.99) 27(75.00) 6(100.00)
下载: 导出CSV
表 4 R778L突变、P992L突变、截断突变的WD患者临床特征分析
Table 4. Analysis of clinical characteristics of WD patients with R778L mutation, P992L mutation and truncating mutation
指标 R778L突变 P992L突变 截断突变 是(n=78) 非(n=85) 是(n=39) 非(n=124) 是(n=32) 非(n=131) 临床信息 男[例(%)] 46(59.0) 48(56.5) 22(56.4) 72(58.1) 16(50.0) 78(59.5) 发病年龄(岁) 8.0 (4.0~13.8) 8.0 (5.0~13.0) 6.0 (4.5~11.0) 8.0
(5.0~15.2)
6.5 (5.0~14.0) 8.0
(4.5~13.0)
肝硬化[例(%)]1) 36(46.2) 39(45.9) 16(41.0) 59(47.6) 15(46.9) 60(45.8) K-F环阳性[例(%)]2) 31/72(43.1) 28/81(34.6) 11/35(31.4) 48/118(40.7) 11/30(36.7) 48/123(39.0) 神经系统异常[例(%)] 3(3.85) 2(2.35) 0(0.00) 5(4.03) 1(3.13) 4(3.05) 肝脏弹性检查3) 8.9(5.7~17.4) 8.5(4.9~19.6) 7.7(5.8~18.4) 9.0(5.1~19.4) 9.1(5.0~18.6) 8.5(5.4~19.2) 实验室检查 INR 1.04(0.98~1.32) 1.04(0.97~1.29) 1.04(0.97~1.29) 1.04(0.98~1.32) 1.04(0.97~1.29) 1.04(0.98~1.32) TBil(μmol/L) 10.3(7.2~23.5) 10.2(7.5~21.3) 9.3(7.6~24.5) 10.9(7.2~20.6) 10.8(7.7~24.5) 10.0(7.3~21.5) ALT(U/L) 103.5 (33.2~199.8) 83.0 (42.0~173.0) 135.0 (80.5~237.0) 80.5 (36.0~173.3) 80.5 (48.8~163.3) 100.0 (40.5~199.5) AST(U/L) 85.0 (50.8~120.3) 84.5 (48.0~144.3) 121.4 (77.0~195.0) 84.0 (39.0~123.3) 87.0 (52.5~126.0) 85.0 (48.0~145.0) 铜生化 24 h尿铜(μg/d)4) 156.0 (99.4~355.0) 155.0 (91.5~403.3) 172.2 (117.9~420.1) 153.0 (87.5~355.3) 160.0 (89.5~605.7) 154.0 (97.3~332.0) 铜蓝蛋白(g/L) 0.04 (0.02~0.08) 0.08 (0.03~0.13) 0.05 (0.04~0.10) 0.05 (0.02~0.11) 0.03 (0.02~0.08) 0.06
(0.03~0.11)
血清铜(mg/L) 3.80 (2.12~6.52) 4.35 (2.70~7.12) 4.30 (2.60~6.85) 3.90 (2.55~7.10) 3.20 (2.15~5.00) 4.20 (2.60~7.50) 注:1)肝硬化包括代偿期肝硬化和失代偿期肝硬化;2)153例患者进行了眼科裂隙灯检查;3)107例患者进行了肝脏弹性检查;4)141例患者检测了24 h尿铜水平。
下载: 导出CSV
-
[1] MALIPATI AEKJ, KAIDIRIYA KEB, XU L, et al. Research advances in the pathogenesis, phenotype-genotype relationship, and pharmacotherapy of hepatolenticular degeneration[J]. J Clin Hepatol, 2023, 39( 6): 1497- 1504. DOI: 10.3969/j.issn.1001-5256.2023.06.037.玛力帕提·艾尔肯江, 凯迪日亚·库尔班, 徐玲, 等. 肝豆状核变性发病机制、临床表型-基因型关系及药物治疗研究进展[J]. 临床肝胆病杂志, 2023, 39( 6): 1497- 1504. DOI: 10.3969/j.issn.1001-5256.2023.06.037. [2] FERENCI P, STREMMEL W, CZŁONKOWSKA A, et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease[J]. Hepatology, 2019, 69( 4): 1464- 1476. DOI: 10.1002/hep.30280. [3] CAI L, HUANG XT, YE Y, et al. Role of gender and age in features of Wilson’s disease[J]. Front Neurol, 2023, 14: 1176946. DOI: 10.3389/fneur.2023.1176946. [4] ROBERTS EA, SCHILSKY ML. Current and emerging issues in Wilson’s disease[J]. N Engl J Med, 2023, 389( 10): 922- 938. DOI: 10.1056/NEJMra1903585. [5] DONG Y, NI W, CHEN WJ, et al. Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilson’s disease guides genetic diagnosis[J]. Theranostics, 2016, 6( 5): 638- 649. DOI: 10.7150/thno.14596. [6] DAS S, MOHAMMED A, MANDAL T, et al. Polarized trafficking and copper transport activity of ATP7B: A mutational approach to establish genotype-phenotype correlation in Wilson disease[J]. Hum Mutat, 2022, 43( 10): 1408- 1429. DOI: 10.1002/humu.24428. [7] ZHANG TH, MAO ZQ. Clinical characteristics and gene analysis of hepatolenticular degeneration in children: A report of 70 cases[J]. Chin J Pract Pediatr, 2022, 37( 2): 135- 139. DOI: 10.19538/j.ek2022020614.张天鹤, 毛志芹. 儿童肝豆状核变性的临床特点及基因分析(附70例报告)[J]. 中国实用儿科杂志, 2022, 37( 2): 135- 139. DOI: 10.19538/j.ek2022020614. [8] XUE ZR, CHEN HY, YU L, et al. A systematic review and meta-analysis of the R778L mutation in ATP7B with Wilson disease in China[J]. Pediatr Neurol, 2023, 145: 135- 147. DOI: 10.1016/j.pediatrneurol.2023.04.026. [9] ZHANG SJ, YANG WM, LI X, et al. Clinical and genetic characterization of a large cohort of patients with Wilson’s disease in China[J]. Transl Neurodegener, 2022, 11( 1): 13. DOI: 10.1186/s40035-022-00287-0. [10] OKADA T, SHIONO Y, KANEKO Y, et al. High prevalence of fulminant hepatic failure among patients with mutant alleles for truncation of ATP7B in Wilson’s disease[J]. Scand J Gastroenterol, 2010, 45( 10): 1232- 1237. DOI: 10.3109/00365521.2010.492527. [11] MERLE U, WEISS KH, EISENBACH C, et al. Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease[J]. BMC Gastroenterol, 2010, 10: 8. DOI: 10.1186/1471-230X-10-8. [12] FERENCI P, CACA K, LOUDIANOS G, et al. Diagnosis and phenotypic classification of Wilson disease[J]. Liver Int, 2003, 23( 3): 139- 142. DOI: 10.1034/j.1600-0676.2003.00824.x. [13] ROZEN S, SKALETSKY H. Primer3 on the WWW for general users and for biologist programmers[J]. Methods Mol Biol, 2000, 132: 365- 386. DOI: 10.1385/1-59259-192-2: 365. [14] JI L, ZHANG Y, KONG L, et al. Clinical features of Wilson’s disease: An analysis of 83 cases[J]. J Clin Hepatol, 2022, 38( 8): 1843- 1846. DOI: 10.3969/j.issn.1001-5256.2022.08.023.纪雷, 张莹, 孔丽, 等. 83例肝豆状核变性患者的临床特征分析[J]. 临床肝胆病杂志, 2022, 38( 8): 1843- 1846. DOI: 10.3969/j.issn.1001-5256.2022.08.023. [15] European Association for study of Liver. EASL clinical practice guidelines: Wilson’s disease[J]. J Hepatol, 2012, 56( 3): 671- 685. DOI: 10.1016/j.jhep.2011.11.007. [16] CHOE EJ, CHOI JW, KANG MJ, et al. A population-based epidemiology of Wilson’s disease in South Korea between 2010 and 2016[J]. Sci Rep, 2020, 10( 1): 14041. DOI: 10.1038/s41598-020-70976-1. [17] OKADA T, SHIONO Y, HAYASHI H, et al. Mutational analysis of ATP7B and genotype-phenotype correlation in Japanese with Wilson’s disease[J]. Hum Mutat, 2000, 15( 5): 454- 462. DOI: 3.0.CO;2-J">10.1002/(SICI)1098-1004(200005)15: 5<454: AID-HUMU7>3.0.CO;2-J. [18] ESPINÓS C, FERENCI P. Are the new genetic tools for diagnosis of Wilson disease helpful in clinical practice?[J]. JHEP Rep, 2020, 2( 4): 100114. DOI: 10.1016/j.jhepr.2020.100114. [19] LIU XQ, ZHANG YF, LIU TT, et al. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease[J]. World J Gastroenterol, 2004, 10( 4): 590- 593. DOI: 10.3748/wjg.v10.i4.590. [20] LALIOTI V, SANDOVAL I, CASSIO D, et al. Molecular pathology of Wilson’s disease: A brief[J]. J Hepatol, 2010, 53( 6): 1151- 1153. DOI: 10.1016/j.jhep.2010.07.008. [21] HUSTER D, HOPPERT M, LUTSENKO S, et al. Defective cellular localization of mutant ATP7B in Wilson’s disease patients and hepatoma cell lines[J]. Gastroenterology, 2003, 124( 2): 335- 345. DOI: 10.1053/gast.2003.50066. [22] FANNI D, GEROSA C, NURCHI VM, et al. Copper-induced epigenetic changes shape the clinical phenotype in Wilson’s disease[J]. Curr Med Chem, 2021, 28( 14): 2707- 2716. DOI: 10.2174/0929867327666200730214757. [23] KIEFFER DA, MEDICI V. Wilson disease: At the crossroads between genetics and epigenetics-a review of the evidence[J]. Liver Res, 2017, 1( 2): 121- 130. DOI: 10.1016/j.livres.2017.08.003. [24] SCHIEFERMEIER M, KOLLEGGER H, MADL C, et al. The impact of apolipoprotein E genotypes on age at onset of symptoms and phenotypic expression in Wilson’s disease[J]. Brain, 2000, 123 Pt 3: 585- 590. DOI: 10.1093/brain/123.3.585. [25] STUEHLER B, REICHERT J, STREMMEL W, et al. Analysis of the human homologue of the canine copper toxicosis gene MURR1 in Wilson disease patients[J]. J Mol Med(Berl), 2004, 82( 9): 629- 634. DOI: 10.1007/s00109-004-0557-9. [26] HUANG YZ, ZHANG M. Advances in molecular mechanism of ATP7B gene mutation in hepatolenticular degeneration[J]. Chin Hepatol, 2023, 28( 7): 866- 872. DOI: 10.14000/j.cnki.issn.1008-1704.2023.07.019.黄元志, 张敏. 肝豆状核变性ATP7B基因突变的分子机制研究进展[J]. 肝脏, 2023, 28( 7): 866- 872. DOI: 10.14000/j.cnki.issn.1008-1704.2023.07.019. -

 本文二维码
本文二维码
计量
- 文章访问数: 1456
- HTML全文浏览量: 719
- PDF下载量: 113
- 被引次数: 0

