
 PDF下载 ( 632 KB)
PDF下载 ( 632 KB)

HBV感染与线粒体稳态相互作用的分子机制
DOI: 10.12449/JCH250222
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:李兴统负责文稿的撰写;周太成负责文稿的审校及修改。
Molecular mechanisms of the interaction between hepatitis B virus infection and mitochondrial homeostasis
-
摘要: HBV感染可造成急性或慢性感染,未经治疗的患者可发展为肝硬化、肝癌而死亡。线粒体作为重要的细胞器之一,其正常形态和功能的维持是保证细胞进行各种生理活动的主要基础,线粒体动力学、线粒体自噬、损伤、氧化磷酸化等生理活动均可影响线粒体稳态的维持。HBV感染可影响线粒体稳态,本文从线粒体动力学、线粒体自噬、线粒体氧化磷酸化及线粒体损伤4个方面总结线粒体稳态与HBV感染的研究进展,深入探讨线粒体稳态的维持与HBV感染的关系,以期为理解HBV感染的分子机制,甚至寻找HBV潜在治疗靶点提供一定的理论依据。Abstract: Hepatitis B virus (HBV) infection can cause acute or chronic infection, while untreated patients can develop into liver cirrhosis or liver cancer, thereby leading to death. As one of the most important organelles of cells, the maintenance of the normal morphology and function of mitochondria is the basis for ensuring various physiological activities in cells, and physiological activities, such as mitochondrial dynamics, mitophagy, injury, and oxidative phosphorylation, can affect the maintenance of mitochondrial homeostasis. HBV infection can affect mitochondrial homeostasis. This article summarizes the research advances in mitochondrial homeostasis and HBV infection from the four aspects of mitochondrial dynamics, mitophagy, mitochondrial oxidative phosphorylation, and mitochondrial injury and discusses the association between the maintenance of mitochondrial homeostasis and HBV infection, in order to provide a theoretical basis for understanding the molecular mechanism of HBV infection and identifying the potential therapeutic targets for HBV.
-
Key words:
- Hepatitis B Virus /
- Mitochondria /
- Mitochondrial Dynamics /
- Mitophagy /
- Oxidative Phosphorylation
-
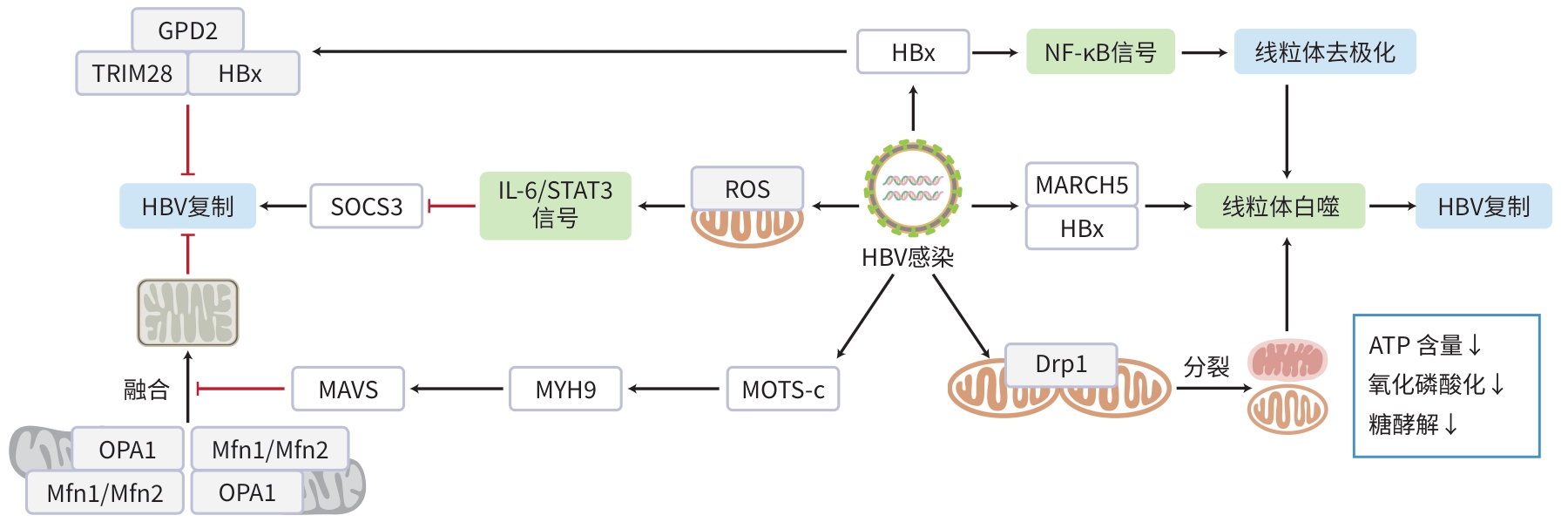
注: ATP,三磷酸腺苷;Drp1,GTP酶动力蛋白相关蛋白1;HBx,HBV X蛋白;GDP,鸟嘌呤二核苷酸磷酸;MAVS,线粒体抗病毒信号蛋白;Mfn,线粒体融合蛋白;MOTS-c,线粒体12S rRNA开放阅读框;MYH9,肌球蛋白-9;OPA1,视神经萎缩1蛋白;ROS,活性氧;SOCS3,细胞因子信号传导3抑制因子;STAT3,信号转导及转录激活因子3;TRIM28,E3泛素连接酶三基序蛋白28。
图 1 HBV感染与线粒体稳态相互作用的分子机制
Figure 1. The molecular mechanisms of the interaction between hepatitis B virus infection and mitochondrial homeostasis.
-
[1] REVILL PA, CHISARI FV, BLOCK JM, et al. A global scientific strategy to cure hepatitis B[J]. Lancet Gastroenterol Hepatol, 2019, 4( 7): 545- 558. DOI: 10.1016/S2468-1253(19)30119-0. [2] SCHWEITZER A, HORN J, MIKOLAJCZYK RT, et al. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013[J]. Lancet, 2015, 386( 10003): 1546- 1555. DOI: 10.1016/S0140-6736(15)61412-X. [3] TSAI WL, CHUNG RT. Viral hepatocarcinogenesis[J]. Oncogene, 2010, 29( 16): 2309- 2324. DOI: 10.1038/onc.2010.36. [4] LIN CR, LUO LJ, XUN Z, et al. Novel function of MOTS-c in mitochondrial remodelling contributes to its antiviral role during HBV infection[J]. Gut, 2024, 73( 2): 338- 349. DOI: 10.1136/gutjnl-2023-330389. [5] WASHIZAKI A, MURAYAMA A, MURATA M, et al. Neutralization of hepatitis B virus with vaccine-escape mutations by hepatitis B vaccine with large-HBs antigen[J]. Nat Commun, 2022, 13( 1): 5207. DOI: 10.1038/s41467-022-32910-z. [6] KIM H, LEE SA, KIM BJ. X region mutations of hepatitis B virus related to clinical severity[J]. World J Gastroenterol, 2016, 22( 24): 5467- 5478. DOI: 10.3748/wjg.v22.i24.5467. [7] LI WH, URBAN S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications[J]. J Hepatol, 2016, 64( 1): S32- S40. DOI: 10.1016/j.jhep.2016.02.011. [8] KOSHIBA T, DETMER SA, KAISER JT, et al. Structural basis of mitochondrial tethering by mitofusin complexes[J]. Science, 2004, 305( 5685): 858- 862. DOI: 10.1126/science.1099793. [9] CAO YL, MENG SX, CHEN Y, et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion[J]. Nature, 2017, 542( 7641): 372- 376. DOI: 10.1038/nature21077. [10] CHAN DC. Mitochondria: Dynamic organelles in disease, aging, and development[J]. Cell, 2006, 125( 7): 1241- 1252. DOI: 10.1016/j.cell.2006.06.010. [11] MATTIE S, RIEMER J, WIDEMAN JG, et al. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space[J]. J Cell Biol, 2018, 217( 2): 507- 515. DOI: 10.1083/jcb.201611194. [12] LOSÓN OC, SONG ZY, CHEN H, et al. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission[J]. Mol Biol Cell, 2013, 24( 5): 659- 667. DOI: 10.1091/mbc.E12-10-0721. [13] INGERMAN E, PERKINS EM, MARINO M, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria[J]. J Cell Biol, 2005, 170( 7): 1021- 1027. DOI: 10.1083/jcb.200506078. [14] JI WK, HATCH AL, MERRILL RA, et al. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites[J]. Elife, 2015, 4: e11553. DOI: 10.7554/eLife.11553. [15] GUDIMCHUK NB, MCINTOSH JR. Regulation of microtubule dynamics, mechanics and function through the growing tip[J]. Nat Rev Mol Cell Biol, 2021, 22( 12): 777- 795. DOI: 10.1038/s41580-021-00399-x. [16] OJAIMI M AL, SALAH A, EL-HATTAB AW. Mitochondrial fission and fusion: Molecular mechanisms, biological functions, and related disorders[J]. Membranes(Basel), 2022, 12( 9): 893. DOI: 10.3390/membranes12090893. [17] CUYÀS E, VERDURA S, MARTIN-CASTILLO B, et al. Circulating levels of MOTS-c in patients with breast cancer treated with metformin[J]. Aging(Albany NY), 2022, 15( 4): 892- 897. DOI: 10.18632/aging.204423. [18] KIM SJ, KHAN M, QUAN J, et al. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis[J]. PLoS Pathog, 2013, 9( 12): e1003722. DOI: 10.1371/journal.ppat.1003722. [19] XIANG L, SHAO YR, CHEN YP. Mitochondrial dysfunction and mito-chondrion-targeted therapeutics in liver diseases[J]. J Drug Target, 2021, 29( 10): 1080- 1093. DOI: 10.1080/1061186X.2021.1909051. [20] SCHOLLMEIER A, BASIC M, GLITSCHER M, et al. The impact of HBx protein on mitochondrial dynamics and associated signaling pathways strongly depends on the hepatitis B virus genotype[J]. J Virol, 2024, 98( 5): e0042424. DOI: 10.1128/jvi.00424-24. [21] SOROURI M, CHANG T, HANCKS DC. Mitochondria and viral infection: Advances and emerging battlefronts[J]. mBio, 2022, 13( 1): e0209621. DOI: 10.1128/mbio.02096-21. [22] SIVASUDHAN E, BLAKE N, LU ZL, et al. Hepatitis B viral protein HBx and the molecular mechanisms modulating the hallmarks of hepatocellular carcinoma: A comprehensive review[J]. Cells, 2022, 11( 4): 741. DOI: 10.3390/cells11040741. [23] LU YY, LI ZJ, ZHANG SQ, et al. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation[J]. Theranostics, 2023, 13( 2): 736- 766. DOI: 10.7150/thno.79876. [24] MATSUDA N, SATO S, SHIBA K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy[J]. J Cell Biol, 2010, 189( 2): 211- 221. DOI: 10.1083/jcb.200910140. [25] PARK S, CHOI SG, YOO SM, et al. Pyruvate stimulates mitophagy via PINK1 stabilization[J]. Cell Signal, 2015, 27( 9): 1824- 1830. DOI: 10.1016/j.cellsig.2015.05.020. [26] NGUYEN TN, PADMAN BS, LAZAROU M. Deciphering the molecular signals of PINK1/parkin mitophagy[J]. Trends Cell Biol, 2016, 26( 10): 733- 744. DOI: 10.1016/j.tcb.2016.05.008. [27] RICHTER B, SLITER DA, HERHAUS L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria[J]. Proc Natl Acad Sci USA, 2016, 113( 15): 4039- 4044. DOI: 10.1073/pnas.1523926113. [28] MOORE AS, HOLZBAUR ELF. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy[J]. Proc Natl Acad Sci USA, 2016, 113( 24): E3349- E3358. DOI: 10.1073/pnas.1523810113. [29] LAZAROU M, SLITER DA, KANE LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy[J]. Nature, 2015, 524( 7565): 309- 314. DOI: 10.1038/nature14893. [30] LI J, YANG DM, LI ZP, et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases[J]. Ageing Res Rev, 2023, 84: 101817. DOI: 10.1016/j.arr.2022.101817. [31] SZARGEL R, SHANI V, ELGHANI F ABD, et al. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway[J]. Hum Mol Genet, 2016, 25( 16): 3476- 3490. DOI: 10.1093/hmg/ddw189. [32] PARK S, CHOI SG, YOO SM, et al. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy[J]. Autophagy, 2014, 10( 11): 1906- 1920. DOI: 10.4161/auto.32177. [33] WONG YC, YSSELSTEIN D, KRAINC D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis[J]. Nature, 2018, 554( 7692): 382- 386. DOI: 10.1038/nature25486. [34] CHENG J, WEI L, LI M. Progress in regulation of mitochondrial dynamics and mitochondrial autophagy[J]. Acta Physiologica Sinica, 2020, 72( 4): 475- 487. DOI: 10.13294/j.aps.2020.0025.程婧, 魏林, 李苗. 线粒体动力学及线粒体自噬调控机制的研究进展[J]. 生理学报, 2020, 72( 4): 475- 487. DOI: 10.13294/j.aps.2020.0025. [35] YAN CJ, GONG LL, CHEN L, et al. PHB2(prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis[J]. Autophagy, 2020, 16( 3): 419- 434. DOI: 10.1080/15548627.2019.1628520. [36] GILKERSON R, KAUR H, CARRILLO O, et al. OMA1-mediated mitochondrial dynamics balance organellar homeostasis upstream of cellular stress responses[J]. Int J Mol Sci, 2024, 25( 8): 4566. DOI: 10.3390/ijms25084566. [37] LIU BH, XU CZ, LIU Y, et al. Mitochondrial quality control in human health and disease[J]. Mil Med Res, 2024, 11( 1): 32. DOI: 10.1186/s40779-024-00536-5. [38] CHEN YY, WANG WH, CHE L, et al. BNIP3L-dependent mitophagy promotes HBx-induced cancer stemness of hepatocellular carcinoma cells via glycolysis metabolism reprogramming[J]. Cancers(Basel), 2020, 12( 3): 655. DOI: 10.3390/cancers12030655. [39] KARBOWSKI M, OSHIMA Y, VERHOEVEN N. Mitochondrial proteotoxicity: Implications and ubiquitin-dependent quality control mechanisms[J]. Cell Mol Life Sci, 2022, 79( 11): 574. DOI: 10.1007/s00018-022-04604-8. [40] YOO YS, PARK YJ, LEE HS, et al. Mitochondria ubiquitin ligase, MARCH5 resolves hepatitis B virus X protein aggregates in the liver pathogenesis[J]. Cell Death Dis, 2019, 10( 12): 938. DOI: 10.1038/s41419-019-2175-z. [41] LIU LJ, LV Z, XUE X, et al. Canonical WNT signaling activated by WNT7B contributes to L-HBs-mediated sorafenib resistance in hepatocellular carcinoma by inhibiting mitophagy[J]. Cancers(Basel), 2022, 14( 23): 5781. DOI: 10.3390/cancers14235781. [42] WING PAC, LIU PJ, HARRIS JM, et al. Hypoxia inducible factors regulate hepatitis B virus replication by activating the basal core promoter[J]. J Hepatol, 2021, 75( 1): 64- 73. DOI: 10.1016/j.jhep.2020.12.034. [43] LIU CY, ZHAO KT, CHEN YS, et al. Mitochondrial glycerol-3-phosphate dehydrogenase restricts HBV replication via the TRIM28-mediated degradation of HBx[J]. J Virol, 2023, 97( 5): e0058023. DOI: 10.1128/jvi.00580-23. [44] SCHMIDT NM, WING PAC, DINIZ MO, et al. Targeting human acyl-CoA: Cholesterol acyltransferase as a dual viral and tcell metabolic checkpoint[J]. Nat Commun, 2021, 12: 2814. DOI: 10.1038/s41467-021-22967-7. [45] LIU Y, XU RY, GU HY, et al. Metabolic reprogramming in macrophage responses[J]. Biomark Res, 2021, 9( 1): 1. DOI: 10.1186/s40364-020-00251-y. [46] LI YM, ZHU YW, FENG S, et al. Macrophages activated by hepatitis B virus have distinct metabolic profiles and suppress the virus via IL-1β to downregulate PPARα and FOXO3[J]. Cell Rep, 2022, 38( 4): 110284. DOI: 10.1016/j.celrep.2021.110284. [47] SUOMALAINEN A, BATTERSBY BJ. Mitochondrial diseases: The contribution of organelle stress responses to pathology[J]. Nat Rev Mol Cell Biol, 2018, 19( 2): 77- 92. DOI: 10.1038/nrm.2017.66. [48] YUAN K, LEI Y, CHEN HN, et al. HBV-induced ROS accumulation promotes hepatocarcinogenesis through Snail-mediated epigenetic silencing of SOCS3[J]. Cell Death Differ, 2016, 23( 4): 616- 627. DOI: 10.1038/cdd.2015.129. [49] CHEN P, YAO LC, YUAN MQ, et al. Mitochondrial dysfunction: A promising therapeutic target for liver diseases[J]. Genes Dis, 2023, 11( 3): 101115. DOI: 10.1016/j.gendis.2023.101115. [50] JABEEN K, JAVED A, MANZOOR S, et al. Antioxidants and calcium modulators preclude in vitro hepatitis B virus-induced mitochondrial damage[J]. Turk J Gastroenterol, 2023, 34( 10): 1052- 1061. DOI: 10.5152/tjg.2023.21290. [51] LIN N, YIN W, MILLER H, et al. The role of regulatory T cells and follicular T helper cells in HBV infection[J]. Front Immunol, 2023, 14: 1169601. DOI: 10.3389/fimmu.2023.1169601. [52] YANG J, ZHENG LY, YANG ZG, et al. 5-FU promotes HBV replication through oxidative stress-induced autophagy dysfunction[J]. Free Radic Biol Med, 2024, 213: 233- 247. DOI: 10.1016/j.freeradbiomed.2024.01.011. [53] YOON H, LEE HK, JANG KL. Hydrogen peroxide inhibits hepatitis B virus replication by downregulating HBx levels via siah-1-mediated proteasomal degradation in human hepatoma cells[J]. Int J Mol Sci, 2023, 24( 17): 13354. DOI: 10.3390/ijms241713354. [54] FENG MX, YU YN, CHEN YQ, et al. HBx-induced PLA2R overexpression mediates podocyte pyroptosis through the ROS-NLRP3 signaling pathway[J]. Ren Fail, 2023, 45( 1): 2170808. DOI: 10.1080/0886022X.2023.2170808. [55] ZHAN X, WU R, KONG XH, et al. Elevated neutrophil extracellular traps by HBV-mediated S100A9-TLR4/RAGE-ROS cascade facilitate the growth and metastasis of hepatocellular carcinoma[J]. Cancer Commun(Lond), 2023, 43( 2): 225- 245. DOI: 10.1002/cac2.12388. -

 下载:
下载:
 本文二维码
本文二维码
图(1)
计量
- 文章访问数: 1134
- HTML全文浏览量: 503
- PDF下载量: 116
- 被引次数: 0

