
 PDF下载 ( 2261 KB)
PDF下载 ( 2261 KB)

铁死亡在胆管癌中的作用机制
DOI: 10.3969/j.issn.1001-5256.2022.04.043
-
摘要: 胆管癌发病率和死亡率不断上升,研究探索新的治疗靶点具有重要意义。铁死亡是一种新型的铁依赖性细胞氧化损伤的细胞死亡方式,与癌症中的铁代谢及氧化应激失衡密切相关,已成为肿瘤领域的研究热点。介绍了铁死亡相关机制,并在此基础上分析铁死亡参与胆管癌发生发展的研究进展,阐明探讨铁死亡相关调控机制在胆管癌恶性进展过程中的作用具有重要临床价值。Abstract: The incidence and mortality rates of cholangiocarcinoma (CCA) are increasing constantly, and it is of great importance to explore new therapeutic targets. Ferroptosis, a unique pattern of cell death caused by iron-dependent cellular oxidative injury, is closely associated with iron metabolism and oxidative stress imbalance in cancer and has become a research hotspot in the field of tumor. This article introduces the mechanism of ferroptosis and the research advances in ferroptosis involved in the development and progression of CCA, and it is pointed out that the regulatory mechanism of ferroptosis has an important clinical value in the malignant progression of CCA.
-
Key words:
- Bile Duct Neoplasms /
- Ferroptosis /
- Pathologic Processes
-
1. 铁死亡的发现及特征
铁死亡是Stocicwell于2012年发现的一种新型细胞死亡方式。在鉴定对Ras突变细胞有致死作用的新分子的研究中,Erastin和RSL3分子以非凋亡方式选择性杀伤细胞,并且伴随着线粒体形态的异常[1]。传统的调控细胞死亡的抑制剂无法阻断此过程,但抗氧化剂(维生素E)和铁螯合剂却可以逆转此过程[2],这种以脂质过氧化为特征的铁依赖性非凋亡性细胞死亡即铁死亡[3]。
与己经鉴定出的11种受调控的细胞死亡方式相比,铁死亡在形态、生化学特征、调控蛋白和功能机制方面都各不相同。譬如,凋亡的主要形态学特征包括细胞质浓缩,染色质高度凝聚,核裂解为碎块,凋亡小体形成,线粒体形态在凋亡早期可维持正常,随着凋亡过程进展可能会出现线粒体肿胀、空泡变等一些非特异性的改变。生化学特征主要有半胱天冬蛋白酶的激活、DNA降解等[4]。自噬的主要形态学特征包括细胞膜特征化结构消失,不发生染色质凝集,出现大量自噬空泡,自噬溶酶体形成,生化学特征主要表现为LC3蛋白-I到LC3蛋白-II的转化、底物降解等[5]。而铁死亡的主要形态学特征表现为细胞核正常,线粒体膜密度升高,线粒体嵴减少;生化学特征主要包括铁和活性氧超载、丝裂原活化蛋白激酶的激活、抑制胱氨酸/谷氨酸逆向转运蛋白系统(cystine/glutamate antiporter system Xc-)和减少半胱氨酸摄取、谷胱甘肽(glutathione,GSH)耗竭、释放花生四烯酸介质等[6]。
2. 铁死亡分子机制
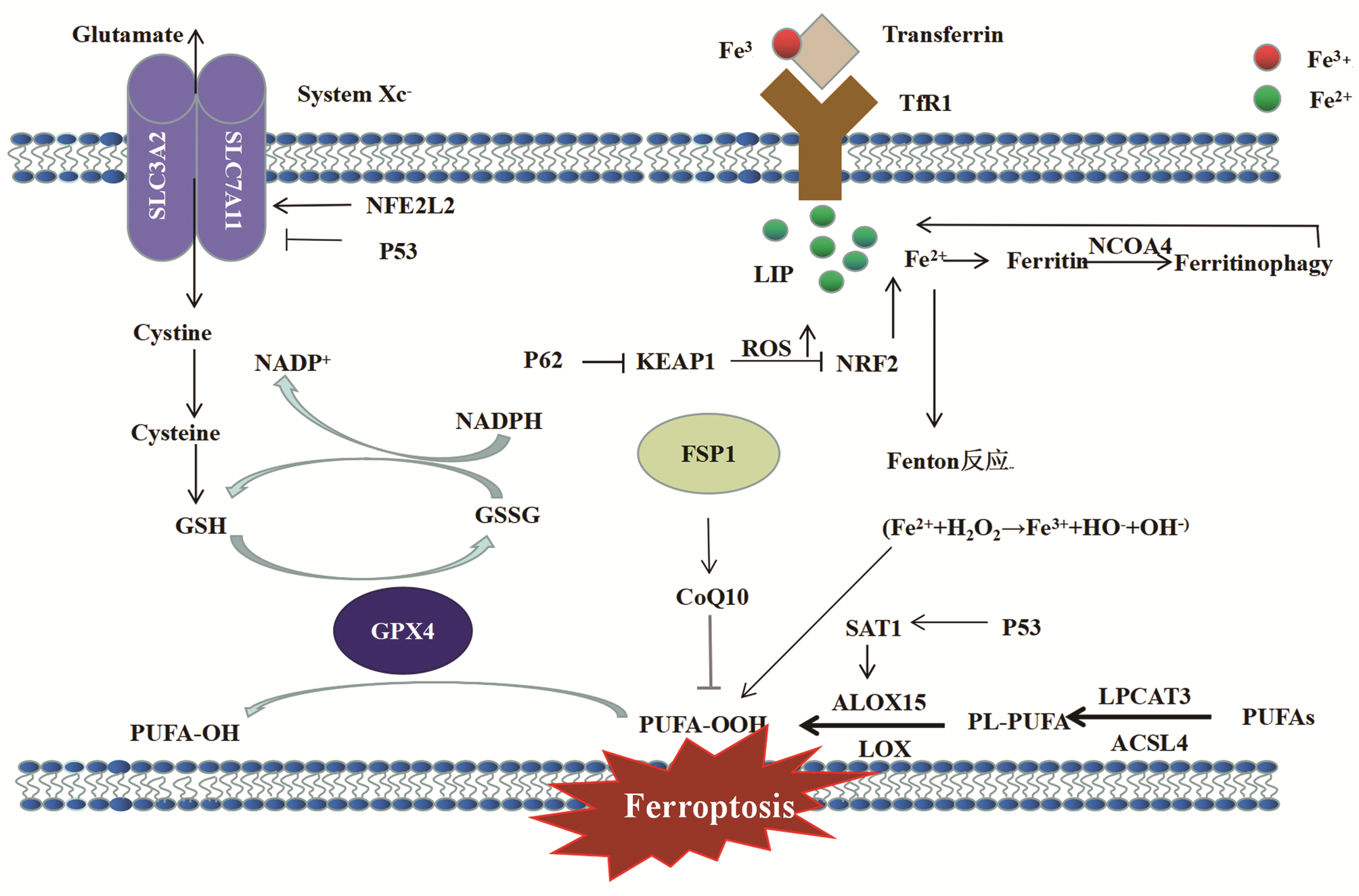
铁死亡是铁依赖性、以脂质活性氧异常增多为特征的一种独立细胞死亡类型。脂质过氧化物的升高和谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)的降低是其发生的两大标志,铁死亡的发生涉及脂质代谢、活性氧的生成与清除、铁离子代谢异常等等,因而其内在分子机制与脂质过氧化、活性氧、GPX4、铁代谢等必然有着密不可分的关系(图 1)。
 图 1 细胞铁死亡调控机制示意图注:ACSL4,长链酰基辅酶a合成酶家族成员4; ALOX-15,花生四烯酸-15脂氧合酶; CoQ10,辅酶Q10; Cysteine,半胱氨酸; Cystine,胱氨酸; Ferritin,铁蛋白; Ferritinophage,铁自噬; FSP1,铁死亡调控蛋白1; Glutamate,谷氨酸; GSSG,氧化型谷胱甘肽; LIP,不稳定铁池; LOX,脂氧合酶; LPCAT3,溶血磷脂酰胆碱酰基转移酶3; NADPH,还原型烟酰胺腺嘌呤二核苷酸磷酸; NFE2L2,红细胞衍生核因子2样蛋白2; NRF2,核因子E2相关因子2; PUFA,多不饱和脂肪酸; SAT1,精胺N1-乙酰转移酶1; SLC3A2,溶质载体家族3成员2; SLC7A11,溶质载体家族7成员11; Transferrin,转铁蛋白; TfR1,转铁蛋白受体1。Figure 1. The schematic diagram of ferroptosis regulation mechanism
图 1 细胞铁死亡调控机制示意图注:ACSL4,长链酰基辅酶a合成酶家族成员4; ALOX-15,花生四烯酸-15脂氧合酶; CoQ10,辅酶Q10; Cysteine,半胱氨酸; Cystine,胱氨酸; Ferritin,铁蛋白; Ferritinophage,铁自噬; FSP1,铁死亡调控蛋白1; Glutamate,谷氨酸; GSSG,氧化型谷胱甘肽; LIP,不稳定铁池; LOX,脂氧合酶; LPCAT3,溶血磷脂酰胆碱酰基转移酶3; NADPH,还原型烟酰胺腺嘌呤二核苷酸磷酸; NFE2L2,红细胞衍生核因子2样蛋白2; NRF2,核因子E2相关因子2; PUFA,多不饱和脂肪酸; SAT1,精胺N1-乙酰转移酶1; SLC3A2,溶质载体家族3成员2; SLC7A11,溶质载体家族7成员11; Transferrin,转铁蛋白; TfR1,转铁蛋白受体1。Figure 1. The schematic diagram of ferroptosis regulation mechanism2.1 活性氧和脂质过氧化的驱动
活性氧在组织内稳态中发挥重要作用,调节细胞信号传导、分化,促进细胞损伤和死亡,其水平受细胞抗氧化系统的严格调控。铁死亡过程中,组织细胞处于氧化应激状态,过量的活性氧产生,攻击生物膜,影响其流动性及结构,通过脂质过氧化链反应,质膜的过氧化反应在膜磷脂之间传递下去,对细胞产生致死作用。但生物体内存在着众多的抗氧化剂,一般的内源性抗氧化系统由酶促抗氧化剂(超氧化物歧化酶、过氧化氢酶、GPX和重组抗氧化酶)和非酶抗氧化剂(维生素或其类似物、矿物质和代谢物)组成,过量的活性氧通过酶促(脂氧合酶催化)和非酶(铁依赖)途径诱导脂质过氧化,导致细胞死亡。
PUFA是脂质过氧化的首选底物,特别是花生四烯酸和肾上腺酸最容易发生过氧化[7-8],导致脂质双分子层被破坏,影响膜功能。细胞膜中PUFA的生物合成和重构需要长链酰基辅酶a合成酶家族成员4(long-chain acyl-CoA synthetases 4, ACSL4)和溶血磷脂酰胆碱酰基转移酶3 (lysophosphatidylcholine acyltransferase 3,LPCAT3)参与[9]。ACSL4在体内可以催化脂肪酸活化合成脂酰辅酶A,这是脂肪酸分解代谢第一步反应的关键酶,而且在脂质过氧化物抑制GPX4的过程中ACSL4是必不可少的。降低ACSL4和LPCAT3的表达可以减少脂质过氧化底物在细胞中的积累,从而抑制铁死亡,因此,ACSL4和LPCAT3已被认为是介导铁死亡的关键蛋白[7]。
2.2 GSH-GPX4
抗氧化剂通过对活性氧等的清除在预防细胞损伤或癌变方面发挥作用。GSH是哺乳动物体内最主要、含量最丰富的含巯基的低分子多肽,有氧化型(GSSG)和还原型(GSH) 两种形式,它可以通过抑制脂质过氧化,清除自由基, 对细胞起到保护作用,是人类细胞内主要的抗氧化剂,也是GPX4的理想底物。半胱氨酸是细胞GSH生物合成中的限速因子,通常在半胱氨酸饥饿,GSH耗竭和抑制GPX4后诱导细胞铁死亡。
GPX4是谷胱甘肽过氧化物酶家族中极其重要的成员。GPX不仅具有清除自由基和衍生物的作用,还减少脂质过氧化物的形成,增强机体抗氧化损伤的能力,从而抑制致癌物和活性氧诱导的肿瘤发生,尤其GPX4可通过抑制脂质过氧化和铁死亡而发挥其重要作用。GPX4基因的缺失可以导致早期胚胎致死。为了确保膜的完整性和减少活性氧引起的损害,GPX4利用还原GSH作为辅助因子,将脂质过氧化氢(R-OOH)转化为脂质醇(R-OH),阻止了Fe2+依赖的毒性活性氧的形成和积累,保护细胞膜中的的多不饱和脂肪酸防止其脂质过氧化[10]。同时细胞内氧化应激状态也可以诱导GPX4活性增高,这可能是与激活核转录因子NF-kB,调节锰-超氧化物歧化酶、血红素加氧酶等抗氧化物质的基因表达相关[11]。GPX4也是铁死亡的关键调节因子,增强GPX4活性以及诱导其表达的因素均会降低细胞对铁死亡的敏感性,反之亦然。研究[12-14]表明,Erastin和RSL3可以分别通过间接和直接的方式抑制GPX4的活性来诱导铁死亡。Xc-系统是异二聚体,由糖基化的重链SLC3A2和非糖基化的SLC7A11通过二硫键连接形成,其中SLC7A11是调控GPX4活性的关键蛋白,SLC7A11-GSH-GPX4是经典的铁死亡通路。SLC7A11的表达和活性进一步受到NFE2L2的正向调节[15],NFE2L2信号通路是抵抗铁死亡的重要防御机制,受到抑癌基因TP53、BRCA1关联蛋白1和Beclin1的负向调控[13]。Xc-系统的抑制决定了GSH水平的下降和铁死亡的开始。而且Xc-转导途径是半胱氨酸合成GSH和维持细胞内硫醇氧化还原电位的重要来源,因此系统Xc-可能是一个很好的抗癌靶点,这也为肿瘤的治疗和降低耐药风险提供了新的思路。此外,铁死亡诱导剂(ferroptosis—inducing agents,FIN)56对GPX4水平也有着显著影响。RSL3可以直接抑制GPX4的活性,而FIN56可降低GPX4的丰度和辅酶Q10的衍生量。FIN56并非Xc-系统的抑制剂,不影响GSH水平,而是通过翻译后降解导致GPX4蛋白的丢失[16]。
2.3 铁代谢
正常的铁对生物的生存至关重要,是许多关键过程中必需元素,主要参与氧运输、DNA生物合成,以及参与形成各种酶,如LOX、黄嘌呤氧化酶、还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate, NADPH)氧化酶、线粒体复合体Ⅰ和Ⅲ等[17]。细胞内铁离子超载,Fe2+可以通过Fenton反应催化形成具有代谢毒性的活性氧和过氧化磷脂,导致细胞损伤或死亡,因此,细胞内铁离子摄入与释放、储存和转运都是影响细胞铁死亡的重要因素[18]。在大鼠中敲减铁离子代谢的关键调节因子——铁反应元件结合蛋白2可以有效降低细胞对铁死亡的敏感性[1]。自噬亦可以通过影响铁离子代谢,进而调节细胞对铁死亡诱导剂的敏感性。储铁蛋白介导的选择性自噬,简称为铁自噬,通过调控细胞内的铁离子转运来增强细胞对铁死亡的敏感性。近期研究[19-20]报道了核因子E2相关因子2 (nuclear factor erythroid2 related factor 2,NRF2)、热休克蛋白B1等基因可通过调节细胞的铁离子代谢,影响细胞对铁死亡诱导剂的敏感性。铁螯合剂可以抑制Erastin诱导的铁死亡,而补充外源性铁则可以增强Erastin诱导的铁死亡。因此,铁离子代谢也是诱导细胞发生铁死亡的重要潜在调控点。
此外,肿瘤细胞的生长往往表现较强的铁依赖性,众多肿瘤调控因子,如Ras、p53、Nrf2等均可以影响铁代谢从而调节肿瘤细胞的死亡。Ras突变的铁死亡细胞与耐铁死亡细胞相比,TfR1表达增加,铁蛋白轻链(ferritin light chain,FTL)和铁蛋白重链1 (ferritin heavy polypeptide1,FTH1)表达降低[21],提示铁摄取增加和铁储存减少可能导致铁死亡过程中的铁过载。铁依赖型铁死亡的更多直接证据也包括来自铁代谢主要转录因子IREB2的研究,通过RNAi抑制IREB2可显著增加铁代谢相关基因的表达(如F-box、FTH1和FTL),因此,靶向铁死亡治疗肿瘤具有临床可行性及重要意义。
2.4 铁死亡其他相关机制及信号通路
2.4.1 P53
P53是与肿瘤关系密切的抑癌基因,也是一种转录因子,通过与靶基因的启动子结合,继而激活或抑制其mRNA的合成[7]。TP53在大约50%的人类癌症中存在双等位突变或缺失,导致肿瘤进展失控。2012年Li等发现p53的翻译后修饰对转录和抑癌作用有重大影响,被称为p533KR的乙酰化缺陷突变体无法诱导凋亡和细胞周期阻滞,但缺乏乙酰化的p53突变体可促进铁死亡[3, 13]。而且,p53是Xc-亚基SCL7A11的编码基因,下调SLC7A11的表达,抑制Xc-系统,从而影响GPX4的活性[13],导致脂质过氧化积累和铁死亡。p53亦可以激活SAT1,调控ALOX-15,促进细胞脂质过氧化,活性氧堆积,最终诱导铁死亡[22],敲除SAT1可显著逆转p53介导的铁死亡。其他一些代谢相关基因,如铁氧还蛋白还原酶[23]和谷氨酰胺酶2[24]等,均是p53介导铁死亡的直接靶点[10],故而p53作为代谢相关基因的调节因子在铁死亡中有着相当重要的地位。
2.4.2 P62-Keap1-Nrf2通路
Keap1-Nrf2系统作为氧化应激、细胞损伤的传感器发挥着重要作用,调控抗氧化酶相关基因的表达[25]。Nrf2是调控氧化还原平衡重要转录因子,在非激活状态下位于细胞质中与Keap1相互作用,并经由泛素-蛋白酶体通路迅速降解。当活性氧水平升高时,Keap1无法泛素化Nrf2,导致Nrf2在细胞核积累,进而诱导与抗氧化、代谢和解毒酶相关的核靶基因表达,这一过程受到自噬受体p62的严格控制,p62是一种多功能蛋白,直接抑制Keap1,同时促进Nrf2激活[26]。研究发现,当肝癌细胞暴露于Erastin或索拉非尼时,p62-keap1-Nrf2通路的激活可阻止Nrf2降解,促进p62核积累,从而抑制铁死亡[20]。
2.4.3 FSP1-CoQ10-NAD(P)H通路
GPX4是抑制铁死亡发生机制的核心,然而研究发现GPX4缺失的细胞仍然能在铁死亡激活状态下生存,在此基础上发现了之前并未识别的铁死亡调控蛋白(ferroptosis suppressor protein 1,FSP1),进一步的研究证明FSP1对铁死亡的抑制是由NAD(P)H依赖辅酶Q10介导,FSP1保护细胞的作用正是通过催化CoQ10的持续再生,提高自由基捕获能力来抑制铁死亡的发生[27-28],对GPX4缺失引起的铁死亡具有保护作用。而且靶向FSP1与GPX4抑制剂具有显著的协同作用,可在多种肿瘤中诱导铁死亡。FSP1-CoQ10- NAD(P)H通路作为一个独立的平行系统,可与GSH-GPX4协同阻遏磷脂过氧化和铁死亡。
除此之外,三羧酸循环和电子传递链,以及Erastin的另一个直接靶点,线粒体电压依赖性阴离子通道[29]都可影响铁死亡。
3. 胆管癌
胆管癌是起源于肝内、门周或远端(肝外)胆道系统的恶性肿瘤,不包括胆囊癌和Vater壶腹癌。2017年,美国癌症联合会和国际抗癌联盟联合发布最新版癌症分期手册,重新定义了门周段胆管癌(perihilor cholangiocarcinoma, pCCA)、远端胆管癌(distal cholangiocarcinoma, dCCA)和肝内胆管癌(intrahepatic cholangiocarcinoma, iCCA)分期系统,罕见的混合性肝细胞-胆管细胞癌被归为iCCA。胆管癌在不同国家的发病率差异很大,多数国家发病率<6例/10万人,但在智利、韩国和泰国北部等一些国家和地区的发病率异常升高[30]。
胆管癌潜伏期较长,早期症状不明显,如肝外胆管细胞癌(extrahepatic cholangiocarcinoma,eCCA)通常在肿瘤阻塞胆道引流系统时才出现症状。多数患者初次就诊时已经处于肿瘤进展期,甚至已发生转移,手术切除和肝移植是3种亚型早期疾病的潜在治愈性治疗选择。然而,由于晚期肿瘤局部浸润,或腹膜、远处转移,或胆道重建选择受限等原因,只有不到1/3的患者在诊断时被归类为可切除的肿瘤手术,并且术后出现局部复发及远处转移高达60%。iCCA切除后的5年生存率为22%~44%,pCCA为11%~41%,dCCA为27%~37%[30]。对于晚期或不能切除的胆管癌患者,吉西他滨联合顺铂是目前常用的一线化疗方案,但中位总生存期<1年[31]。此外,用于CCA早期诊断、预后预测和治疗评价的生物标志物仍相当有限[32],如CA19-9和CEA是目前研究相对充分的两种标志物,但其结果与良性疾病及其他恶性肿瘤存在明显重叠,诊断价值有限,而且他们对早期胆管细胞癌的敏感性较低,因此有必要深入探究关键分子机制,寻找新的诊治靶点。
研究报道胆管癌发病的诱因包括肝内胆管结石、原发性硬化性胆管炎、肝吸虫病、病毒性肝炎以及先天性胆道畸形等多种危险因素,但其共同特征是多数都会导致胆汁淤积,进而引起胆管的炎性病变,氧化应激失衡,直至癌症发生。此外,在胆管癌发生过程中铁代谢异常也参与其中,伴随着铁依赖性氧化应激增强[32],因此,胆管癌恶性进展过程中的氧化应激失衡与铁代谢异常均提示新型的细胞死亡方式铁死亡参与其中,进一步探讨铁死亡在胆管癌发生发展中的可能机制,对胆管癌的防治具有重要的指导意义。
3.1 胆管癌与氧化应激失衡
诱发胆管癌的危险因素众多,但其共同特点即因胆管的慢性梗阻,导致胆汁淤积,进而引起胆管的慢性炎性。有动物模型研究[33]证实,慢性胆管阻塞可以引起胆管癌的发生与发展。此外,在炎症微环境中,活性氧/活性氮可损伤生物大分子(DNA、蛋白质和脂质),导致其功能障碍,形成氧化应激失衡的恶性循环,诱发癌症。氧化应激的失衡及氧化还原信号通路的失调是癌症进展和对治疗耐药的共同特征[34-35]。近来研究发现,铁离子也参与氧化应激引起的生物大分子损伤过程,炎症诱导的氧化应激可以导致Fe3+结合和TfR氧化,促进铁积累和释放;此外,铁调素的表达通过骨形态发生蛋白和蛋白酪氨酸激酶2/信号转导子和转录激活子3信号通路调控[36]。在炎症或感染刺激后,IL-6激活,通过铁调素依赖性途径或其他途径抑制铁转运蛋白活性[37],从而导致细胞内铁超载,增加宿主对感染的易感性或癌症发生。羰基化是氧化应激诱导的一种不可逆的蛋白质修饰。羰基化蛋白在胆管癌组织中表达显著升高。炎症诱导的血清转铁蛋白、热休克蛋白70和Α1抗胰蛋白酶的羰基化可能在胆管癌的癌变中起关键作用[38]。血清转铁蛋白羰基化,可导致铁的积累和释放,增强Fenton反应。此外,具有抗氧化性质的热休克蛋白70和具有蛋白酶抑制能力的Α1抗胰蛋白酶的羰基化则导致他们功能障碍,降解GPX4,促进铁死亡,从而导致胆管癌的进展和不良预后[38]。综上,炎症反应及相关氧化应激失衡在胆管癌的发生发展中有着至关重要的作用。
3.2 胆管癌与铁代谢
肝脏是铁在体内储存的主要部位,过量的的铁参与Fenton反应,产生活性氧,进而促进多不饱和脂肪酸的过氧化反应,导致严重的氧化应激和细胞损伤。与非肿瘤细胞相比,肿瘤细胞(特别是肿瘤干细胞)的生长具有明显的铁依赖性[7]。肿瘤细胞内铁水平的调节可以概括为:铁摄取增加,铁储存和输出减少,从而铁水平升高以维持肿瘤细胞生长的高铁需求[37]。铁稳态的破坏在肿瘤发生和治疗抵抗中发挥重要作用。
LIP是Fenton反应的铁离子源,Fenton反应产生过量活性氧,参与了胆管癌发生发展的全过程[38]。铁调控蛋白的异常表达或修饰以及TfR-1也在胆管癌发生发展中具有关键作用。上调TfR-1导致铁摄取增强,LIP增加,从而促进肿瘤进展;敲除TfR-1则降低细胞内LIP、抑制胆管癌细胞迁移能力和增殖能力,并且铁超载与胆管癌患者的不良预后显著相关[39]。Tran等[40]发现在104例胆管癌肿瘤组织中,铁蛋白、铁调素和运铁蛋白的表达较相对于周围非癌组织降低,但转铁蛋白受体表达上调,转铁蛋白受体功能障碍可以显著抑制Erastin诱导的铁死亡。此外,暴露于外源性过氧化氢可以激活铁响应元件,通过结合mRNA来抑制翻译或降解铁蛋白,参与调控铁代谢过程[41]。为探索胆管癌治疗的新方向提供了依据。
3.3 其他相关分子及信号通路
异柠檬酸脱氢酶1/2(isocit rate dehydrogenase1/2,IDH1/2),BRCA1关联蛋白1,AT丰富结构域1A和多溴蛋白1是iCCA中最常见的突变基因。IDH1/2突变与胆管癌中p53水平升高、铁代谢和DNA高甲基化显著相关[42],并且与IDH突变相关的表观遗传学变化可能介导了其致癌效应。此外,胆管癌的发生常继发于细胞DNA的损伤,存在多种癌基因与抑癌基因的突变,>21%的病例Kras癌基因与>37%的病例p53抑癌基因出现异常表达,表现为侵袭性更强的基因显型[43]。在胆管癌患者的胆汁与胰液中同样可以发现Kras基因与p53基因的突变,而这两种基因在铁死亡中都发挥重要作用。Kras/ERK信号传导途径下游的ADP-核糖基化因子6蛋白可通过抑制RSL3诱导铁死亡降低对吉西他滨的耐药性[44]。近期研究表明,抑制Xc-系统可以通过诱导铁死亡逆转头颈癌细胞对顺铂的耐药[45],因此,铁死亡的异常调控亦参与了肿瘤细胞的耐药抵抗。
4. 总结与展望
铁死亡是近年来发现的一种新型细胞死亡形式。随着研究的不断深入,发现其内在调控机制涉及多种分子的表达及信号通路。肿瘤细胞中铁代谢异常及氧化应激失衡为通过调控铁死亡靶向治疗肿瘤提供了理论依据。虽然铁死亡在肿瘤的发生、发展及治疗的具体机制尚未完全阐明,但食品药品监督管理局已批准的索拉非尼、柳氮磺胺吡啶等在肿瘤治疗中效果显著[7],而且铁死亡在放疗、免疫治疗、分子靶向治疗、纳米医学抗肿瘤等方面也有明显作用。胆管癌中铁死亡调控机制研究已有一定进展,但仍需要进一步从代谢途径、表观修饰等多个层面深入分析相关机制,推动诊治能力的提升,从而干预疾病的发生发展。
-

注:ACSL4,长链酰基辅酶a合成酶家族成员4; ALOX-15,花生四烯酸-15脂氧合酶; CoQ10,辅酶Q10; Cysteine,半胱氨酸; Cystine,胱氨酸; Ferritin,铁蛋白; Ferritinophage,铁自噬; FSP1,铁死亡调控蛋白1; Glutamate,谷氨酸; GSSG,氧化型谷胱甘肽; LIP,不稳定铁池; LOX,脂氧合酶; LPCAT3,溶血磷脂酰胆碱酰基转移酶3; NADPH,还原型烟酰胺腺嘌呤二核苷酸磷酸; NFE2L2,红细胞衍生核因子2样蛋白2; NRF2,核因子E2相关因子2; PUFA,多不饱和脂肪酸; SAT1,精胺N1-乙酰转移酶1; SLC3A2,溶质载体家族3成员2; SLC7A11,溶质载体家族7成员11; Transferrin,转铁蛋白; TfR1,转铁蛋白受体1。
图 1 细胞铁死亡调控机制示意图
Figure 1. The schematic diagram of ferroptosis regulation mechanism
-
[1] DIXON SJ, LEMBERG KM, LAMPRECHT MR, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death[J]. Cell, 2012, 149(5): 1060-1072. DOI: 10.1016/j.cell.2012.03.042. [2] YAMADA N, KARASAWA T, KIMURA H, et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure[J]. Cell Death Dis, 2020, 11(2): 144. DOI: 10.1038/s41419-020-2334-2. [3] LI J, CAO F, YIN HL, et al. Ferroptosis: Past, present and future[J]. Cell Death Dis, 2020, 11(2): 88. DOI: 10.1038/s41419-020-2298-2. [4] XU X, LAI Y, HUA ZC. Apoptosis and apoptotic body: Disease message and therapeutic target potentials[J]. Biosci Rep, 2019, 39(1): BSR20180992. DOI: 10.1042/BSR20180992. [5] GALLUZZI L, GREEN DR. Autophagy-independent functions of the autophagy machinery[J]. Cell, 2019, 177(7): 1682-1699. DOI: 10.1016/j.cell.2019.05.026. [6] LEE H, ZANDKARIMI F, ZHANG Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis[J]. Nat Cell Biol, 2020, 22(2): 225-234. DOI: 10.1038/s41556-020-0461-8. [7] CHEN X, KANG R, KROEMER G, et al. Broadening horizons: The role of ferroptosis in cancer[J]. Nat Rev Clin Oncol, 2021, 18(5): 280-296. DOI: 10.1038/s41571-020-00462-0. [8] ZHANG FY, ADILA·YKP, ZHAO JM, et al. Mechanism of ferroptosis and its role in liver diseases[J]. J Clin Hepatol, 2021, 37(6): 1454-1458. DOI: 10.3969/j.issn.1001-5256.2021.06.049.张飞宇, 阿迪拉·亚克普, 赵金明, 等. 铁死亡的发生机制及在肝脏疾病中的作用[J]. 临床肝胆病杂志, 2021, 37(6): 1454-1458. DOI: 10.3969/j.issn.1001-5256.2021.06.049. [9] DIXON SJ, WINTER GE, MUSAVI LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death[J]. ACS Chem Biol, 2015, 10(7): 1604-1609. DOI: 10.1021/acschembio.5b00245. [10] FEI W, CHEN D, TANG H, et al. Targeted GSH-exhausting and hydroxyl radical self-producing manganese-silica nanomissiles for MRI guided ferroptotic cancer therapy[J]. Nanoscale, 2020, 12(32): 16738-16754. DOI: 10.1039/d0nr02396e. [11] BANNING A, BRIGELIUS-FLOHÉ R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression[J]. Antioxid Redox Signal, 2005, 7(7-8): 889-899. DOI: 10.1089/ars.2005.7.889. [12] YANG WS, SRIRAMARATNAM R, WELSCH ME, et al. Regulation of ferroptotic cancer cell death by GPX4[J]. Cell, 2014, 156(1-2): 317-331. DOI: 10.1016/j.cell.2013.12.010. [13] JIANG L, KON N, LI T, et al. Ferroptosis as a p53-mediated activity during tumour suppression[J]. Nature, 2015, 520(7545): 57-62. DOI: 10.1038/nature14344. [14] SATO M, KUSUMI R, HAMASHIMA S, et al. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells[J]. Sci Rep, 2018, 8(1): 968. DOI: 10.1038/s41598-018-19213-4. [15] CHEN D, TAVANA O, CHU B, et al. NRF2 is a major target of ARF in p53-independent tumor suppression[J]. Mol Cell, 2017, 68(1): 224-232. e4. DOI: 10.1016/j.molcel.2017.09.009. [16] SHIMADA K, SKOUTA R, KAPLAN A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis[J]. Nat Chem Biol, 2016, 12(7): 497-503. DOI: 10.1038/nchembio.2079. [17] SILVA I, RAUSCH V, PECCERELLA T, et al. Hypoxia enhances H2O2-mediated upregulation of hepcidin: Evidence for NOX4-mediated iron regulation[J]. Redox Biol, 2018, 16: 1-10. DOI: 10.1016/j.redox.2018.02.005. [18] JIA F, SONG N, WANG W, et al. High dietary iron supplement induces the nigrostriatal dopaminergic neurons lesion in transgenic mice expressing mutant A53T human alpha-synuclein[J]. Front Aging Neurosci, 2018, 10: 97. DOI: 10.3389/fnagi.2018.00097. [19] SUN X, OU Z, XIE M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death[J]. Oncogene, 2015, 34(45): 5617-5625. DOI: 10.1038/onc.2015.32. [20] SUN X, OU Z, CHEN R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells[J]. Hepatology, 2016, 63(1): 173-184. DOI: 10.1002/hep.28251. [21] YANG M, LI X, LI H, et al. Baicalein inhibits RLS3-induced ferroptosis in melanocytes[J]. Biochem Biophys Res Commun, 2021, 561: 65-72. DOI: 10.1016/j.bbrc.2021.05.010. [22] ASSAILY W, RUBINGER DA, WHEATON K, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress[J]. Mol Cell, 2011, 44(3): 491-501. DOI: 10.1016/j.molcel.2011.08.038. [23] ZHANG Y, FENG X, ZHANG J, et al. Iron regulatory protein 2 is a suppressor of mutant p53 in tumorigenesis[J]. Oncogene, 2019, 38(35): 6256-6269. DOI: 10.1038/s41388-019-0876-5. [24] CHANG HW, LEE M, LEE YS, et al. p53-dependent glutamine usage determines susceptibility to oxidative stress in radioresistant head and neck cancer cells[J]. Cell Signal, 2021, 77: 109820. DOI: 10.1016/j.cellsig.2020.109820. [25] TSAI TF, CHEN PC, LIN YC, et al. Miconazole contributes to NRF2 activation by noncanonical P62-KEAP1 pathway in bladder cancer cells[J]. Drug Des Devel Ther, 2020, 14: 1209-1218. DOI: 10.2147/DDDT.S227892. [26] SUN Y, HE L, WANG T, et al. Activation of p62-Keap1-Nrf2 pathway protects 6-hydroxydopamine-induced ferroptosis in dopaminergic cells[J]. Mol Neurobiol, 2020, 57(11): 4628-4641. DOI: 10.1007/s12035-020-02049-3. [27] DOLL S, FREITAS FP, SHAH R, et al. FSP1 is a glutathione-independent ferroptosis suppressor[J]. Nature, 2019, 575(7784): 693-698. DOI: 10.1038/s41586-019-1707-0. [28] BERSUKER K, HENDRICKS JM, LI Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis[J]. Nature, 2019, 575(7784): 688-692. DOI: 10.1038/s41586-019-1705-2. [29] GAO M, YI J, ZHU J, et al. Role of mitochondria in ferroptosis[J]. Mol Cell, 2019, 73(2): 354-363. e3. DOI: 10.1016/j.molcel.2018.10.042. [30] BANALES JM, CARDINALE V, CARPINO G, et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA)[J]. Nat Rev Gastroenterol Hepatol, 2016, 13(5): 261-280. DOI: 10.1038/nrgastro.2016.51. [31] WEIGT J, MALFERTHEINER P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer[J]. Expert Rev Gastroenterol Hepatol, 2010, 4(4): 395-397. DOI: 10.1586/egh.10.45. [32] HAN JY, AHN KS, BAEK WK, et al. Usefulness of bile as a biomarker via ferroptosis and cysteine prenylation in cholangiocarcinoma; role of diagnosis and differentiation from benign biliary disease[J]. Surg Oncol, 2020, 34: 174-181. DOI: 10.1016/j.suronc.2020.04.019. [33] MOHR R, ÖZDIRIK B, KNORR J, et al. In vivo models for cholangiocarcinoma-what can we learn for human disease?[J]. Int J Mol Sci, 2020, 21(14): 4993. DOI: 10.3390/ijms21144993. [34] PARK JH, PYUN WY, PARK HW. Cancer metabolism: Phenotype, signaling and therapeutic targets[J]. Cells, 2020, 9(10): 2038. DOI: 10.3390/cells9102308. [35] KIRTONIA A, SETHI G, GARG M. The multifaceted role of reactive oxygen species in tumorigenesis[J]. Cell Mol Life Sci, 2020, 77(22): 4459-4483. DOI: 10.1007/s00018-020-03536-5. [36] GANZ T. Systemic iron homeostasis[J]. Physiol Rev, 2013, 93(4): 1721-1741. DOI: 10.1152/physrev.00008.2013. [37] MANCINELLI R, CUTONE A, ROSA L, et al. Different iron-handling in inflamed small and large cholangiocytes and in small and large-duct type intrahepatic cholangiocarcinoma[J]. Eur J Histochem, 2020, 64(4): 3156. DOI: 10.4081/ejh.2020.3156. [38] THANAN R, OIKAWA S, YONGVANIT P, et al. Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma[J]. Free Radic Biol Med, 2012, 52(8): 1465-1472. DOI: 10.1016/j.freeradbiomed.2012.01.018. [39] JAMNONGKAN W, THANAN R, TECHASEN A, et al. Upregulation of transferrin receptor-1 induces cholangiocarcinoma progression via induction of labile iron pool[J]. Tumour Biol, 2017, 39(7): 1010428317717655. DOI: 10.1177/1010428317717655. [40] TRAN KT, COLEMAN HG, MCCAIN RS, et al. Serum biomarkers of iron status and risk of primary liver cancer: A systematic review and meta-analysis[J]. Nutr Cancer, 2019, 71(8): 1365-1373. DOI: 10.1080/01635581.2019.1609053. [41] CHEN GQ, BENTHANI FA, WU J, et al. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis[J]. Cell Death Differ, 2020, 27(1): 242-254. DOI: 10.1038/s41418-019-0352-3. [42] MA B, MENG H, TIAN Y, et al. Distinct clinical and prognostic implication of IDH1/2 mutation and other most frequent mutations in large duct and small duct subtypes of intrahepatic cholangiocarcinoma[J]. BMC Cancer, 2020, 20(1): 318. DOI: 10.1186/s12885-020-06804-6. [43] NABESHIMA T, HAMADA S, TAGUCHI K, et al. Keap1 deletion accelerates mutant K-ras/p53-driven cholangiocarcinoma[J]. Am J Physiol Gastrointest Liver Physiol, 2020, 318(3): g419-g427. DOI: 10.1152/ajpgi.00296.2019. [44] YE Z, HU Q, ZHUO Q, et al. Abrogation of ARF6 promotes RSL3-induced ferroptosis and mitigates gemcitabine resistance in pancreatic cancer cells[J]. Am J Cancer Res, 2020, 10(4): 1182-1193. [45] LEE J, YOU JH, SHIN D, et al. Inhibition of glutaredoxin 5 predisposes cisplatin-resistant head and neck cancer cells to ferroptosis[J]. Theranostics, 2020, 10(17): 7775-7786. DOI: 10.7150/thno.46903. 期刊类型引用(4)
1. 曹健,董钦鹏,曾炼,李恒平,刘君瑞,孙晓东,王青松,胡鹏超. 褪黑素调控自噬及铁死亡增强胰腺癌细胞PANC-1对吉西他滨的化疗敏感性. 医药导报. 2024(04): 502-510 .  百度学术
百度学术2. 申文龙,李世朋,李泽信,王迎,王亮,王建国. 沉默NCOA4表达对胆管癌细胞增殖、迁移与侵袭的影响. 中国老年学杂志. 2024(15): 3744-3749 . 百度学术3. 景瑞花,乔鑫利,薛菲. 白内障氧化应激损伤诱导晶状体上皮细胞铁死亡. 眼科新进展. 2023(04): 274-278 . 百度学术4. 张瑞鹏,李杰. lncRNA GPRC5D-AS1对地塞米松诱导小鼠成肌细胞肌萎缩的抵抗和再生作用及其机制. 吉林大学学报(医学版). 2023(06): 1457-1465 . 百度学术其他类型引用(1)
-

 下载:
下载:
 下载:
下载:
 百度学术
百度学术 本文二维码
本文二维码
计量
- 文章访问数: 1333
- HTML全文浏览量: 307
- PDF下载量: 132
- 被引次数: 5

