
 PDF下载 ( 2214 KB)
PDF下载 ( 2214 KB)

LPL基因变异导致的婴儿家族性高乳糜微粒血症合并肾脏错构瘤1例报告
DOI: 10.3969/j.issn.1001-5256.2023.04.022
伦理学声明: 本例报告通过复旦大学附属儿科医院伦理委员会批准, 批号: 复儿伦审(2020)402号,已获得患者家属知情同意。
利益冲突声明: 所有作者均声明不存在利益冲突。
作者贡献声明: 陈欣涛、方微园负责文章的构思与设计,数据收集整理,资料分析,撰写论文, 贡献相当, 排名不分先后; 龚晓妍参与数据分析和解释; 林琼、陆怡负责拟定写作思路, 指导撰写文章, 质量控制及审校。
Infantile familial chylomicronemia syndrome caused by LPL gene variants coexisting with renal hamartoma:A case report
-
-
关键词:
- 脂蛋白脂酶缺乏症Ⅰ型 /
- 高甘油三酯血症 /
- 肾脏错构瘤
-
Key words:
- Hyperlipoproteinemia Type I /
- Hypertriglyceridemia /
- Renal Hamartoma
-

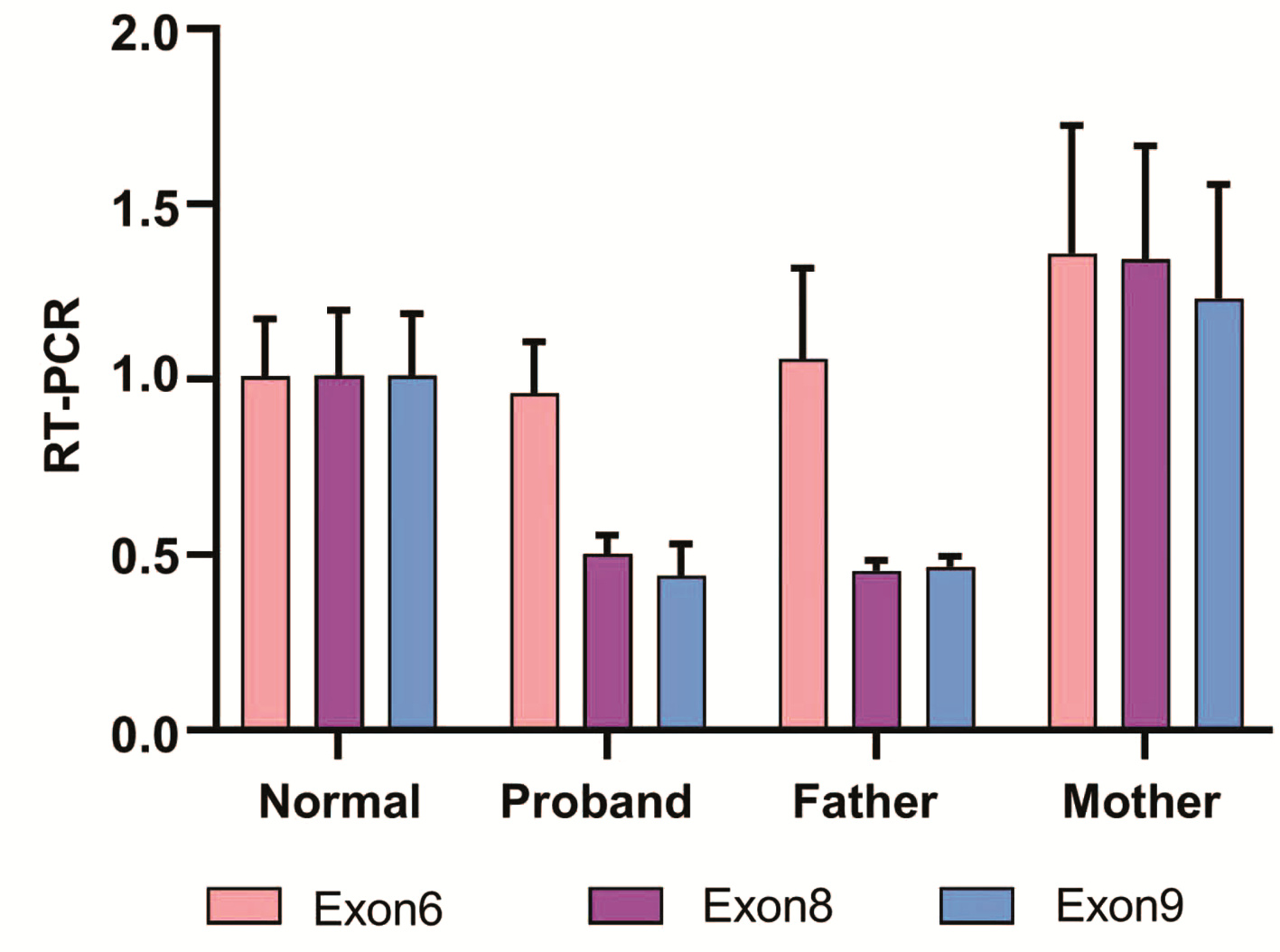
图 1 LPL基因Sanger测序结果
注:a, 患儿; b, 患儿母亲;c, 患儿父亲。
Figure 1. Sanger sequencing result of LPL gene
-
[1] YANG L, DONG XR, PENG XM, et al. Evaluation of turn around time and diagnostic accuracy of the next generation sequencing data analysis pipeline version 2 of Children's Hospital of Fudan University[J]. Chin J Evid Based Pediatr, 2018, 13(2): 118-123. DOI: 10.3969/j.issn.1673-5501.2018.02.008.杨琳, 董欣然, 彭小敏, 等. 复旦大学附属儿科医院高通量测序数据分析流程(第二版)对遗传疾病候选变异基因筛选用时和准确性分析[J]. 中国循证儿科杂志, 2018, 13(2): 118-123. DOI: 10.3969/j.issn.1673-5501.2018.02.008 [2] QIN Q, LIU B, YANG L, et al. Application of copy number variation screening analysis process based on high-throughput sequencing technology[J]. Chin J Evid Based Pediatr, 2018, 13(4): 275-279. DOI: 10.3969/j.issn.1673-5501.2018.04.007.秦谦, 刘博, 杨琳, 等. 基于高通量测序技术的拷贝数变异筛选分析流程的建立及应用[J]. 中国循证儿科杂志, 2018, 13(4): 275-279. DOI: 10.3969/j.issn.1673-5501.2018.04.007. [3] KERSTEN S. Physiological regulation of lipoprotein lipase[J]. Biochim Biophys Acta, 2014, 1841(7): 919-933. DOI: 10.1016/j.bbalip.2014.03.013. [4] OLIVECRONA G. Role of lipoprotein lipase in lipid metabolism[J]. Curr Opin Lipidol, 2016, 27(3): 233-241. DOI: 10.1097/MOL.0000000000000297. [5] BRAHM A, HEGELE RA. Hypertriglyceridemia[J]. Nutrients, 2013, 5(3): 981-1001. DOI: 10.3390/nu5030981. [6] DRON JS, HEGELE RA. Genetics of hypertriglyceridemia[J]. Front Endocrinol (Lausanne), 2020, 11: 455. DOI: 10.3389/fendo.2020.00455. [7] FALKO JM. Familial chylomicronemia syndrome: A clinical guide for endocrinologists[J]. Endocr Pract, 2018, 24(8): 756-763. DOI: 10.4158/EP-2018-0157. [8] TAKAGI A, IKEDA Y. Genetic diagnosis on hypertriglyceridemia-analysis for LPL gene mutations[J]. Nihon Rinsho, 2013, 71(9): 1569-1576. [9] GARG A, GARG V, HEGELE RA, et al. Practical definitions of severe versus familial hypercholesterolaemia and hypertriglyceridaemia for adult clinical practice[J]. Lancet Diabetes Endocrinol, 2019, 7(11): 880-886. DOI: 10.1016/S2213-8587(19)30156-1. [10] LIU LX, WU YT. Clinical advances in hypertriglyceridemia[J]. Chin J Evidence-Bases Cardiovascular Med, 2020, 12(4): 504-508. DOI: 10.3969/j.issn.1674-4055.2020.04.34.刘立新, 武云涛. 高甘油三酯血症临床新进展[J]. 中国循证心血管医学杂志, 2020, 12(4): 504-508. DOI: 10.3969/j.issn.1674-4055.2020.04.34. [11] CHYZHYK V, BROWN AS. Familial chylomicronemia syndrome: A rare but devastating autosomal recessive disorder characterized by refractory hypertriglyceridemia and recurrent pancreatitis[J]. Trends Cardiovasc Med, 2020, 30(2): 80-85. DOI: 10.1016/j.tcm.2019.03.001. [12] GALLO A, BÉLIARD S, D'ERASMO L, et al. Familial chylomicronemia syndrome (FCS): recent data on diagnosis and treatment[J]. Curr Atheroscler Rep, 2020, 22(11): 63. DOI: 10.1007/s11883-020-00885-1. [13] MOULIN P, DUFOUR R, AVERNA M, et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an "FCS score"[J]. Atherosclerosis, 2018, 275: 265-272. DOI: 10.1016/j.atherosclerosis.2018.06.814. [14] VALAIYAPATHI B, SUNIL B, ASHRAF AP. Approach to hypertriglyceridemia in the pediatric population[J]. Pediatr Rev, 2017, 38(9): 424-434. DOI: 10.1542/pir.2016-0138. [15] CALIÒ A, BRUNELLI M, SEGALA D, et al. Angiomyolipoma of the kidney: from simple hamartoma to complex tumour[J]. Pathology, 2021, 53(1): 129-140. DOI: 10.1016/j.pathol.2020.08.008. [16] CHENG L, GU J, EBLE JN, et al. Molecular genetic evidence for different clonal origin of components of human renal angiomyolipomas[J]. Am J Surg Pathol, 2001, 25(10): 1231-1236. DOI: 10.1097/00000478-200110000-00002. [17] PFIRMANN P, COMBE C, RIGOTHIER C. Tuberous sclerosis complex: A review[J]. Rev Med Interne, 2021, 42(10): 714-721. DOI: 10.1016/j.revmed.2021.03.003. [18] CURATOLO P, BOMBARDIERI R, JOZWIAK S. Tuberous sclerosis[J]. Lancet, 2008, 372(9639): 657-668. DOI: 10.1016/S0140-6736(08)61279-9. [19] GILBERT B, ROUIS M, GRIGLIO S, et al. Lipoprotein lipase (LPL) deficiency: a new patient homozygote for the preponderant mutation Gly188Glu in the human LPL gene and review of reported mutations: 75% are clustered in exons 5 and 6[J]. Ann Genet, 2001, 44(1): 25-32. DOI: 10.1016/s0003-3995(01)01037-1. [20] OKAZAKI H, GOTODA T, OGURA M, et al. Current diagnosis and management of primary chylomicronemia[J]. J Atheroscler Thromb, 2021, 28(9): 883-904. DOI: 10.5551/jat.RV17054. [21] STEINHAGEN-THIESSEN E, STROES E, SORAN H, et al. The role of registries in rare genetic lipid disorders: Review and introduction of the first global registry in lipoprotein lipase deficiency[J]. Atherosclerosis, 2017, 262: 146-153. DOI: 10.1016/j.atherosclerosis.2016.08.023. [22] WITZTUM JL, GAUDET D, FREEDMAN SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome[J]. N Engl J Med, 2019, 381(6): 531-542. DOI: 10.1056/NEJMoa1715944. [23] ELKINS C, FRUH S, JONES L, et al. Clinical practice recommendations for pediatric dyslipidemia[J]. J Pediatr Health Care, 2019, 33(4): 494-504. DOI: 10.1016/j.pedhc.2019.02.009. [24] CHAUDHRY R, VILJOEN A, WIERZBICKI AS. Pharmacological treatment options for severe hypertriglyceridemia and familial chylomicronemia syndrome[J]. Expert Rev Clin Pharmacol, 2018, 11(6): 589-598. DOI: 10.1080/17512433.2018.1480368. [25] MEYERS CD, TREMBLAY K, AMER A, et al. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome[J]. Lipids Health Dis, 2015, 14: 8. DOI: 10.1186/s12944-015-0006-5. -

 下载:
下载:
 本文二维码
本文二维码
图(2)
计量
- 文章访问数: 420
- HTML全文浏览量: 99
- PDF下载量: 30
- 被引次数: 0

