
 PDF下载 ( 1926 KB)
PDF下载 ( 1926 KB)

91例以单纯性黄疸为表现的不明原因肝病患者病因谱及临床特征分析
DOI: 10.3969/j.issn.1001-5256.2023.05.016
Etiological spectrum and clinical features of patients with unexplained liver disease manifesting as isolated jaundice: An analysis of 91 cases
-
摘要:
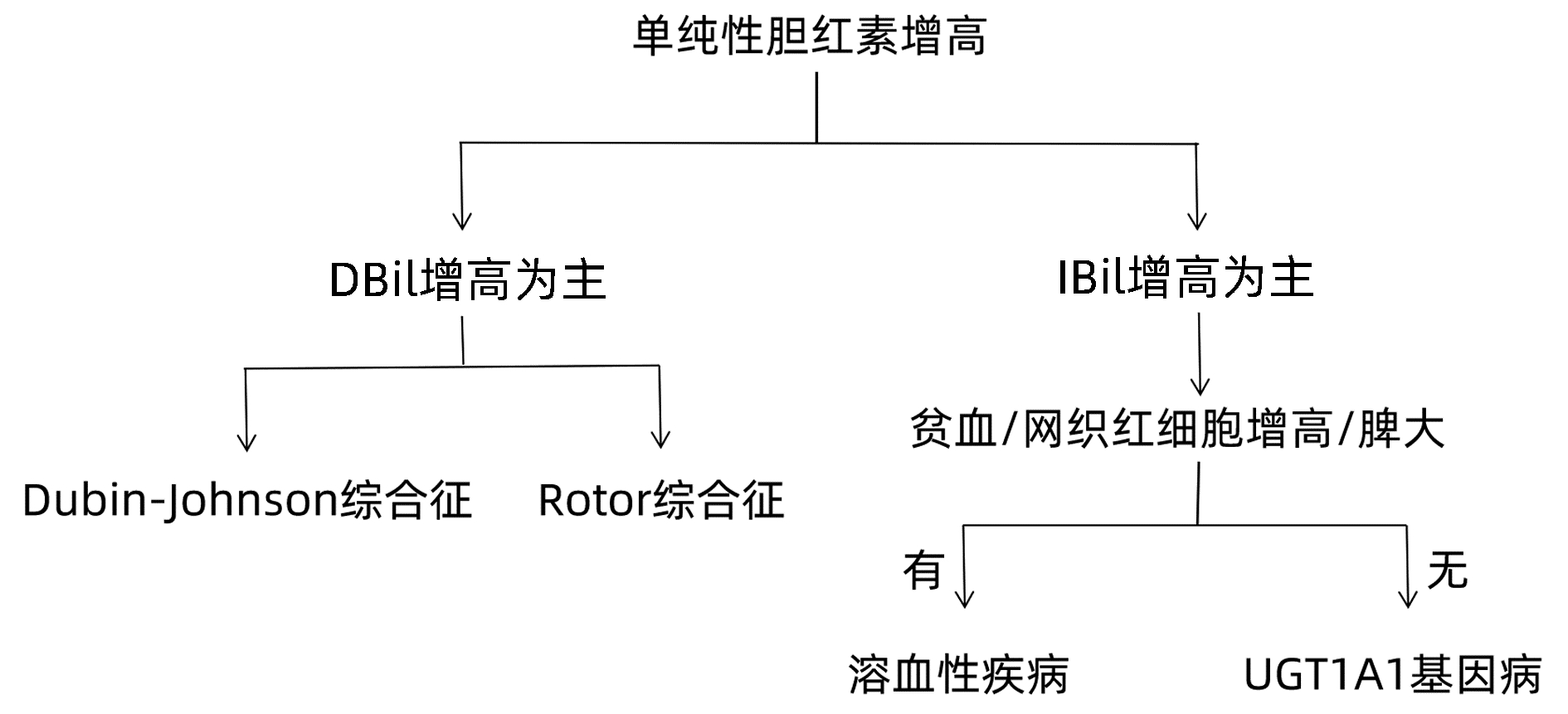
目的 分析表现为单纯性黄疸的不明原因肝病患者的病因和临床特征,以及全外显子测序对该类疾病的诊断价值。 方法 回顾性分析2017年2月—2021年12月因不明原因肝病于南京市第二医院就诊并且进行全外显子基因测序患者的临床资料,根据肝功能指标及影像学资料将所有病例进行临床表型归类,结合全外显子基因测序报告作出诊断。非正态分布的计量资料两组间比较采用Mann-Whitney U检验,多组间比较采用Kruskal-Wallis H检验。 结果 全外显子基因测序病例共519例,排除临床资料缺失或不完整病例102例,纳入417例患者,其中以单纯性黄疸为表现的患者有91例(21.82%,91/417)。通过全外显子基因测序仍无法明确黄疸升高病因者8例(8.79%,8/91)。结合基因检测结果报告分析,83例(91.21%,83/91)患者明确诊断,其中包括遗传性高胆红素血症68例(74.73%,68/91)、遗传性球形红细胞增多症3例(3.30%,3/91)、丙酮酸激酶缺乏症2例(2.20%,2/91)以及UGT1A1基因病合并其他疾病10例(10/91,10.99%)。遗传性高胆红素血症为主要病因,其中UGT1A1基因病61例(67.03%,61/91)、Dubin-Johnson综合征5例(5.49%,5/91)、Rotor综合征2例(2.20%,2/91)。以上各类诊断患者IBil/TBil水平存在显著性差异(H=22.835,P=0.001),UGT1A1基因病合并其他疾病患者的TBil水平明显高于单纯性UGT1A1基因病患者[95.8(37.5~187.1) μmol/L vs 51.4(34.8~267.1) μmol/L,Z=-2.372,P=0.018]。 结论 通过全外显子基因测序可诊断大部分表现为单纯性黄疸的不明原因肝病,其中遗传性高胆红素血症为主要病因,以UGT1A1基因病最为多见。对于表现为单纯性黄疸的不明原因肝病,全外显子基因测序技术可辅助临床诊断。 Abstract:Objective To investigate the etiological and clinical features of patients with unexplained liver disease manifesting as isolated jaundice and the value of whole-exome sequencing in the diagnosis of such diseases. Methods A retrospective analysis was performed for the clinical data of the patients who attended Nanjing Second Hospital due to unexplained liver disease and underwent whole-exome sequencing from February 2017 to December 2021, and according to liver function parameters and imaging data, all cases were classified based on clinical phenotype and were diagnosed based on the whole-exome sequencing report. The Mann-Whitney U test was used for comparison of non-normally distributed continuous data between two groups, and the Kruskal-Wallis H test was used for comparison between multiple groups. Results A total of 519 patients underwent whole-exome sequencing, among whom 102 patients with missing or incomplete clinical data were excluded, and finally 417 patients were included in analysis, among whom 91(91/417, 21.82%) had the manifestation of isolated jaundice. The etiology of jaundice was not determined by whole-exome sequencing in 8 patients (8/91, 8.79%). With reference to genetic testing results, 83 patients (83/91, 91.21%) had a confirmed diagnosis, among whom there were 68 patients with hereditary hyperbilirubinemia (68/91, 74.72%), 3 patients with hereditary spherocytosis (3/91, 3.30%), 2 patients with pyruvate kinase deficiency (2/91, 2.20%), and 10 patients with UGT1A1 gene disease combined with other diseases (10/91, 10.99%). Hereditary hyperbilirubinemia was the main etiology, and there were 61 patients with UGT1A1 gene disease (61/91, 67.03%), 5 patients with Dubin-Johnson syndrome (5/91, 5.49%) and 2 patients with Rotor syndrome (2/91, 2.20%). There was a significant difference in indirect bilirubin/total bilirubin ratio between the patients with the different diagnoses above (H=22.835, P < 0.05), and the patients with UGT1A1 gene disease and other diseases had a significantly higher level of total bilirubin than those with UGT1A1 gene disease alone [95.8 (37.5-187.1) μmol/L vs 51.4 (34.8-267.1) μmol/L, Z=-2.372, P=0.018]. Conclusion Whole-exome sequencing can help with the diagnosis of most cases of unexplained liver disease manifesting as isolated jaundice. Hereditary hyperbilirubinemia is the main etiology, and UGT1A1 gene disease is the most common disease. Whole-exome sequencing can assist the clinical diagnosis of unexplained liver disease manifesting as isolated jaundice. -
Key words:
- Liver Diseases /
- Jaundice /
- Diagnosis /
- Exome
-
表 1 91例单纯性黄疸不明原因肝病患者疾病诊断构成、性别及年龄分布
Table 1. Diagnostic composition, sex and age distribution of 91 patients with isolated jaundice of unknown cause of liver disease
诊断 例数 男/女(例) 年龄范围(岁) 平均年龄(岁) 遗传性高胆红素血症 UGT1A1基因病 61 45/16 17~58 37.06±12.42 DJS 5 3/2 15~59 28.20±18.20 RS 2 1/1 19~31 25.00±8.49 溶血性疾病 HS 3 2/1 16~49 37.33±18.50 PKD 2 2/0 44~48 46.00±2.83 UGT1A1基因病合并其他疾病 合并DJS 4 3/1 17~58 37.25±22.82 合并ADPKD 1 1/0 23 23 合并RS 1 1/0 32 32 合并HS 3 1/2 33~53 40.33±11.02 合并CDA-2 1 1/0 46 46 未明确诊断 8 6/2 14~64 33.63±15.28 注:DJS,Dubin-Johnson综合征;RS,Rotor综合征;HS,遗传性球形红细胞增多症;PKD,丙酮酸激酶缺乏症;ADPKD,常染色体显性多囊肾病;CDA-2,先天性红细胞生成异常性贫血Ⅱ型。  下载: 导出CSV
下载: 导出CSV
表 2 单纯性黄疸各类诊断病例胆红素水平
Table 2. Bilirubin of each diagnosis in isolated jaundice cases
诊断 例数 TBil(μmol/L) DBil(μmol/L) IBil(μmol/L) 遗传性高胆红素血症 UGT1A1基因病 61 51.4(34.8~267.1) 11.1(3.4~19.5) 39.8(22.8~248.1) DJS 5 60.0(40.0~89.0) 44.5(28.3~54.8) 15.0(11.7~38.1) RS 2 46.9(43.8~50.0) 28.1(22.5~33.6) 18.9(16.4~21.3) 溶血性疾病 HS 3 77.2(39.9~107.4) 9.2(7.5~21.6) 68.0(32.4~85.8) PKD 2 52.1(49.2~55.0) 13.0(12.8~13.2) 39.1(36.0~42.3) UGT1A1基因病合并其他疾病 合并DJS/RS 5 108.1(39.2~187.1) 95.9(37.2~146.7) 27.2(2.0~64.1) 合并HS 3 78.3(37.5~103.3) 7.4(6.9~7.8) 70.9(29.7~96.4) 合并CDA-2 1 140.6 10.1 130.5 合并ADPKD 1 64.9 22.7 42.2 未明确诊断 8 54.7(39.1~114.1) 13.6(7.3~101.2) 31.7(12.9~75.3)
下载: 导出CSV
表 3 单纯性UGT1A1基因病与UGT1A1基因病合并其他疾病胆红素比较
Table 3. Comparison of bilirubin between isolated UGT1A1 gene disease and UGT1A1 gene disease combined with other diseases
指标 UGT1A1基因病(n=61) UGT1A1基因病合并其他疾病(n=10) Z值 P值 TBil(μmol/L) 51.4(34.8~267.1) 95.8(37.5~187.1) -2.372 0.018 DBil(μmol/L) 11.1(3.4~19.5) 30.0(6.9~146.7) -0.926 0.355 IBil(μmol/L) 39.8(22.8~248.1) 41.3(2.0~130.5) -0.132 0.895
下载: 导出CSV
表 4 各类诊断IBil/TBil水平比较
Table 4. Comparison of IBil/TBil ratio among all kinds of disease
组别 例数 IBil/TBil UGT1A1基因病 61 0.78(0.60~0.94) DJS 5 0.28(0.19~0.43) RS 2 0.41(0.33~0.49) HS 3 0.81(0.80~0.88) PKD 2 0.75(0.73~0.77) UGT1A1基因病合并其他疾病 10 0.69(0.05~0.93) 未明确诊断 8 0.69(0.11~0.91)
下载: 导出CSV
-
[1] YANG YF. Road map of diagnosis in patients with unknown causes[J]. J Prac Hepatol, 2018, 21(1): 1-3. DOI: 10.3969/j.issn.1672-5069.2018.01.001.杨永峰. 不明原因肝病诊断思路[J]. 实用肝脏病杂志, 2018, 21(1): 1-3. DOI: 10.3969/j.issn.1672-5069.2018.01.001. [2] MEMON N, WEINBERGER BI, HEGYI T, et al. Inherited disorders of bilirubin clearance[J]. Pediatr Res, 2016, 79(3): 378-386. DOI: 10.1038/pr.2015.247. [3] STRASSBURG CP. Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome)[J]. Best Pract Res Clin Gastroenterol, 2010, 24(5): 555-571. DOI: 10.1016/j.bpg.2010.07.007. [4] MARUO Y, BEHNAM M, IKUSHIRO S, et al. Two different UGT1A1 mutations causing Crigler-Najjar syndrome types I and Ⅱ in an Iranian family[J]. J Gastrointestin Liver Dis, 2015, 24(4): 523-526. DOI: 10.15403/jgld.2014.1121.244.ugt. [5] BAI J, ZHENG SJ, DUAN ZP. Clinical features and diagnosis of four common types of congenital hyperbilirubinemia[J]. J Clin Hepatol, 2019, 35(8): 1680-1683. DOI: 10.3969/j.issn.1001-5256.2019.08.005.白洁, 郑素军, 段钟平. 4种常见先天性高胆红素血症的临床特征及诊断思路[J]. 临床肝胆病杂志, 2019, 35(8): 1680-1683. DOI: 10.3969/j.issn.1001-5256.2019.08.005. [6] DHAWAN A, LAWLOR MW, MAZARIEGOS GV, et al. Disease burden of Crigler-Najjar syndrome: Systematic review and future perspectives[J]. J Gastroenterol Hepatol, 2020, 35(4): 530-543. DOI: 10.1111/jgh.14853. [7] KRINGEN MK, PIEHLER AP, GRIMHOLT RM, et al. Serum bilirubin concentration in healthy adult North-Europeans is strictly controlled by the UGT1A1 TA-repeat variants[J]. PLoS One, 2014, 9(2): e90248. DOI: 10.1371/journal.pone.0090248. [8] ERLINGER S, ARIAS IM, DHUMEAUX D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences[J]. Gastroenterology, 2014, 146(7): 1625-1638. DOI: 10.1053/j.gastro.2014.03.047. [9] CHRISTOFORIDOU A, FASOULAKIS Z, KONTOMANOLIS EN. Diagnosis and management of congenital dyserythropoietic anaemia Type Ⅱ in a secundigravida[J]. Cureus, 2017, 9(10): e1811. DOI: 10.7759/cureus.1811. [10] BERGMANN C, GUAY-WOODFORD LM, HARRIS PC, et al. Polycystic kidney disease[J]. Nat Rev Dis Primers, 2018, 4(1): 50. DOI: 10.1038/s41572-018-0047-y. [11] BOTSTEIN D, RISCH N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease[J]. Nat Genet, 2003, 33 Suppl: 228-237. DOI: 10.1038/ng1090. -

 本文二维码
本文二维码
计量
- 文章访问数: 487
- HTML全文浏览量: 106
- PDF下载量: 82
- 被引次数: 0

