
 PDF下载 ( 2397 KB)
PDF下载 ( 2397 KB)

锚蛋白重复序列22(ANKRD22)对人肝癌细胞的影响及其机制
DOI: 10.12449/JCH240519
Effect of ankyrin-repeat domain-containing protein 22 on human hepatoma cells and its mechanism
-
摘要:
目的 探讨锚蛋白重复序列22(ANKRD22)对人肝癌细胞增殖、侵袭和迁移的影响及其分子机制。 方法 通过TCGA数据库分析正常肝组织及肝细胞癌组织中ANKRD22的表达水平及其与预后的关系。通过qRT-PCR和Western Blot检测人正常肝细胞(L-02)和人肝癌细胞系(Huh7、Hep G2、MHCC-97H、SK-HEP-1、SMMC-7721)中ANKRD22的表达情况。通过CCK-8、EdU、划痕实验及Transwell检测ANKRD22对肝癌细胞增殖、侵袭和迁移能力的影响。通过Western Blot检测ANKRD22与细胞周期蛋白、EMT相关蛋白之间的关系。通过KEGG、ssGSEA分析进一步探究ANKRD22在肝癌细胞中的作用机制,并进行实验验证。计量资料两组间比较采用成组t检验,多组间比较采用单因素方差分析,进一步两两比较采用LSD-t检验。 结果 TCGA数据库中ANKRD22在肝细胞癌组织中较正常肝组织高表达(t=5.083,P<0.05),且ANKRD22高表达患者的总生存期及疾病相关生存期均显著低于ANKRD22低表达的患者(P值均<0.05)。肝癌细胞系中ANKRD22的表达量均高于正常肝细胞(P值均<0.05)。增殖实验结果提示,ANKRD22过表达组的EdU阳性率、增殖速度均高于空载对照(Vector)组(t值分别为19.60、6.72,P值均<0.001);si-ANKRD22#2组及si-ANKRD22#3组的EdU阳性率、增殖速度较si-NC组均降低(P值均<0.001)。Cyclin E1、Cyclin D1、CDK7、CDK4在过表达组中表达高于Vector组(t值分别为3.54、4.95、6.34、5.19,P值均<0.01);在si-ANKRD22#2组及si-ANKRD22#3组中表达均低于si-NC组(P值均<0.001)。P27在过表达组中表达低于Vector组(t=6.12,P<0.001),在si-ANKRD22#2组及si-ANKRD22#3组中表达均高于si-NC组(P值均<0.001)。侵袭、迁移实验结果提示,ANKRD22过表达组的迁移速度、穿膜数量(迁移组和侵袭组)均高于Vector组(t值分别为5.01、25.60、3.67,P值均<0.05);si-ANKRD22#2组及si-ANKRD22#3组的迁移速度、穿膜数量(迁移组和侵袭组)较si-NC组均降低(P值均<0.01)。N-cadherin、Vimentin、Snail在过表达组中表达高于Vector组(t值分别为12.13、8.85、13.97,P值均<0.001),在si-ANKRD22#2组及si-ANKRD22#3组中表达均低于si-NC组(P值均<0.001);E-cadherin在过表达组中表达低于Vector组(t=4.98,P<0.01),在si-ANKRD22#2组及si-ANKRD22#3组中表达均高于si-NC组(P值均<0.001)。KEGG富集分析及ssGSEA分析提示,ANKRD22在肝细胞癌中与PI3K/AKT/mTOR信号通路相关,在过表达组中,p-AKT/AKT、p-PI3K/PI3K、p-mTOR/mTOR均较Vector组升高(t值分别为12.21、3.43、9.75,P值均<0.01);在si-ANKRD22#2组及si-ANKRD22#3组中表达均低于si-NC组(P值均<0.001)。 结论 ANKRD22在肝癌细胞中高表达,能促进肝癌细胞的增殖、侵袭和迁移能力,并且能促进PI3K/AKT/mTOR信号通路的激活。 Abstract:Objective To investigate the effect of ankyrin-repeat domain-containing protein 22 (ANKRD22) on the proliferation, invasion, and migration of human hepatoma cells and its molecular mechanism. Methods The TCGA database was used to analyze the expression level of ANKRD22 in normal liver tissue and hepatocellular carcinoma tissue and its association with prognosis. Western Blot and qRT-PCR were used to measure the expression of ANKRD22 in human normal liver cells (L-02) and human hepatoma cells (Huh7, HepG2, MHCC-97H, SK-HEP-1, and SMMC-7721); CCK-8 assay, EdU, wound healing assay, and Transwell assay were used to observe the effect of ANKRD22 on the proliferation, invasion, and migration of hepatoma cells; Western Blot was used to investigate the association of ANKRD22 with cyclins and EMT-related proteins; KEGG and ssGSEA analyses were performed to investigate the mechanism of action of ANKRD22 in hepatoma cells, and related experiments were conducted for validation. The independent-samples t-test was used for comparison of continuous data between two groups; a one-way analysis of variance was used for comparison between multiple groups, and the least significant difference t-test was used for further comparison between two groups. Results In the TCGA database, the expression level of ANKRD22 in hepatoma tissue was significantly higher than that in normal liver tissue (t=5.083, P<0.05), and the patients with a high expression level of ANKRD22 had longer overall survival and disease-related survival than those with a low expression level of ANKRD22 (P<0.05). The expression level of ANKRD22 in various human hepatoma cell lines was higher than that in human normal liver cells (all P<0.05). Cell proliferation assay showed that the ANKRD22 overexpression group had significantly higher EdU positive rate and proliferation rate than the Vector group (t=19.60 and 6.72, both P<0.001), and compared with the si-NC group, the si-ANKRD22#2 group and the si-ANKRD22#3 group had significantly lower EdU positive rate and proliferation rate (all P<0.001). Compared with the Vector group, the overexpression group had significantly higher expression levels of Cyclin E1, Cyclin D1, CDK7, and CDK4 (t=3.54, 4.95, 6.34, and 5.19, all P<0.01), and the si-ANKRD22#2 group and the si-ANKRD22#3 group had significantly lower expression levels than the si-NC group (all P<0.001). The overexpression group had a significantly lower expression level of P27 than the Vector group (t=6.12, P<0.001), and the si-ANKRD22#2 group and the si-ANKRD22#3 group had a significantly higher expression level than the si-NC group (both P<0.001). Invasion and migration experiments showed that compared with the Vector group, the ANKRD22 overexpression group had significantly higher migration rate and number of crossings through the membrane (migration group and invasion group) (t=5.01, 25.60, and 3.67, all P<0.05), and compared with the si-NC group, thesi-ANKRD22#2 group and the si-ANKRD22#3 group had significantly lower migration rate and number of crossings through the membrane (migration group and invasion group) (all P<0.01). The overexpression group had significantly higher expression levels of N-cadherin, Vimentin, and Snail than the Vector group (t=12.13, 8.85, and 13.97, all P<0.001), and the si-ANKRD22#2 group and the si-ANKRD22#3 group had significantly lower expression levels than the si-NC group (all P<0.001); the overexpression group had a significantly lower expression level of E-cadherin than the Vector group (t=4.98, P<0.01), and the si-ANKRD22#2 group and the si-ANKRD22#3 group had a significantly higher expression level than the si-NC group (both P<0.001). The KEGG enrichment analysis and the ssGSEA analysis showed that ANKRD22 was associated with the PI3K/AKT/mTOR signaling pathway in hepatocellular carcinoma, and the overexpression group had significantly higher expression levels of p-AKT/AKT, p-PI3K/PI3K, and p-mTOR/mTOR than the Vector group (t=12.21, 3.43, and 9.75, all P<0.01), and the si-ANKRD22#2 group and the si-ANKRD22#3 group had significantly lower expression levels than the si-NC group (all P<0.001). Conclusion ANKRD22 is highly expressed in hepatoma cells and can promote the proliferation, invasion, and migration of hepatoma cells and the activation of the PI3K/AKT/mTOR signaling pathway. -
Key words:
- Carcinoma, Hepatocellular /
- Ankyrin Repeat /
- Cell Proliferation /
- Cell Movement
-
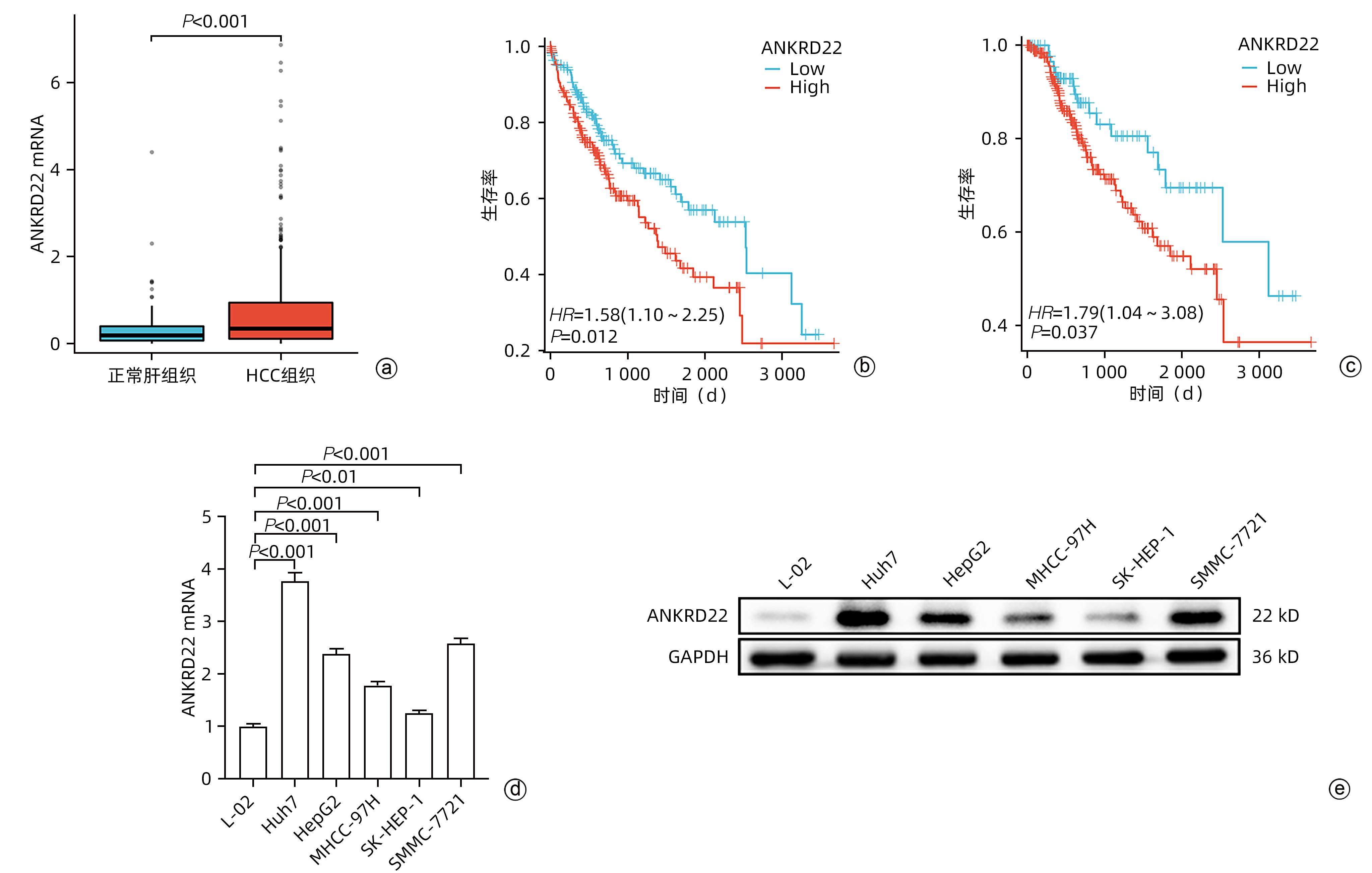
注: a,ANKRD22在正常肝组织与HCC组织中的表达水平;b,ANKRD22不同表达水平患者的总生存期;c,ANKRD22不同表达水平患者的疾病相关生存期;d,ANKRD22在正常肝细胞与肝癌细胞系中的mRNA水平;e,ANKRD22在正常肝细胞与肝癌细胞系中的蛋白表达水平。
图 1 ANKRD22在HCC中的表达情况
Figure 1. Expression of ANKRD22 in hepatocellular carcinoma
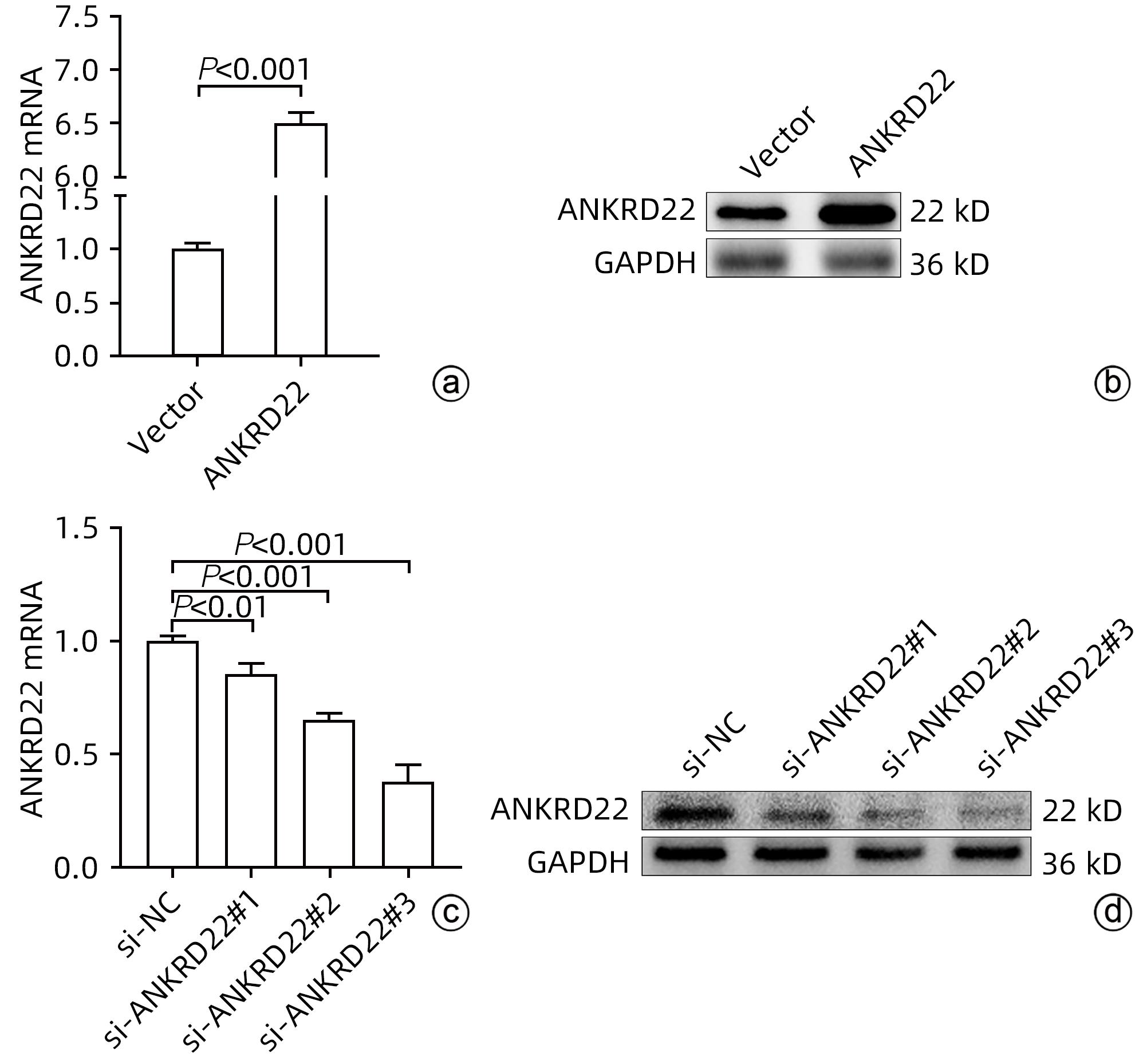
注: a,ANKRD22在Vector组及过表达组MHCC-97H中的mRNA水平;b,ANKRD22在Vector组及过表达组MHCC-97H中的蛋白表达水平;c,ANKRD22在si-NC组及各敲低组Huh7中的mRNA水平;d,ANKRD22在si-NC组及各敲低组Huh7中的蛋白表达水平。
图 2 ANKRD22转染后的表达情况
Figure 2. Expression of ANKRD22 after transfection
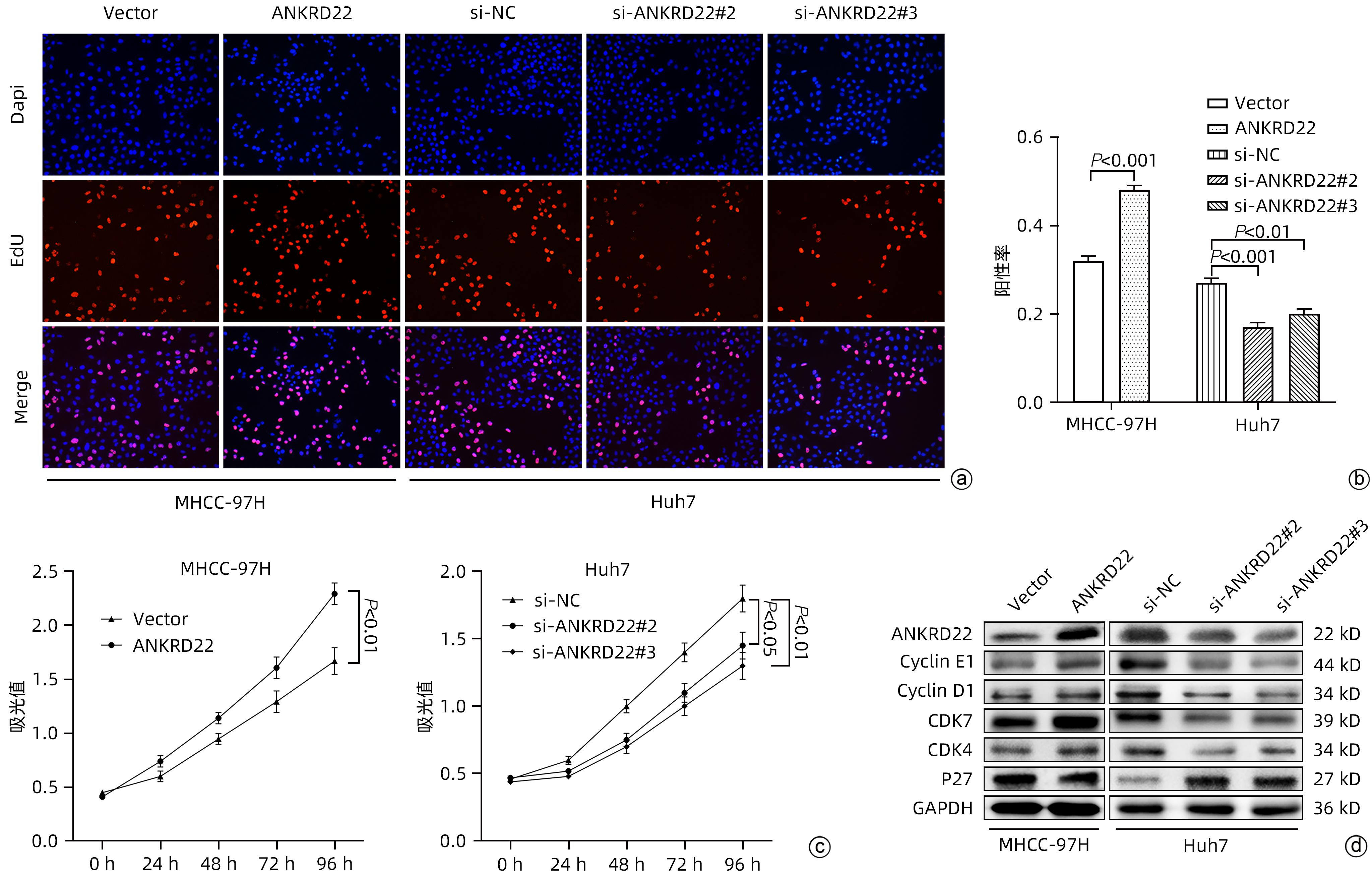
注: a,EdU增殖试验结果;b,EdU阳性率统计结果;c,CCK-8试验统计结果;d,各分组中周期蛋白表达情况。
图 3 ANKRD22对肝癌细胞增殖能力的影响
Figure 3. Effect of ANKRD22 on proliferation of hepatocellular carcinoma cells

注: a,划痕实验结果;b,划痕处迁移面积; c,Transwell实验结果;d,Transwell迁移细胞数;e,Transwell侵袭细胞数;f,各分组中EMT相关蛋白表达情况。
图 4 ANKRD22对肝癌细胞侵袭、迁移能力的影响
Figure 4. Effect of ANKRD22 on invasion and migration of hepatocellular carcinoma cells
-
[1] LLOVET JM, KELLEY RK, VILLANUEVA A, et al. Hepatocellular carcinoma[J]. Nat Rev Dis Primers, 2021, 7( 1): 6. DOI: 10.1038/s41572-020-00240-3. [2] FOGLIA B, TURATO C, CANNITO S. Hepatocellular carcinoma: Latest research in pathogenesis, detection and treatment[J]. Int J Mol Sci, 2023, 24( 15): 12224. DOI: 10.3390/ijms241512224. [3] DING CM, HOU JF, TAO GW, et al. Early diagnosis and screening of hepatocellular carcinoma[J/OL]. Chin J Hepatic Surg Electron Ed, 2023, 12( 1): 22- 28. DOI: 10.3877/cma.j.issn.2095-3232.2023.01.005.丁成明, 侯嘉丰, 陶光伟, 等. 肝细胞癌早期诊断和筛查[J/OL]. 中华肝脏外科手术学电子杂志, 2023, 12( 1): 22- 28. DOI: 10.3877/cma.j.issn.2095-3232.2023.01.005. [4] KIM E, VIATOUR P. Hepatocellular carcinoma: Old friends and new tricks[J]. Exp Mol Med, 2020, 52( 12): 1898- 1907. DOI: 10.1038/s12276-020-00527-1. [5] LIU XF, ZHANG J, YAO L, et al. Advances in targeted therapy combined with immunotherapy for advanced hepatocellular carcinoma[J]. J Clin Hepatol, 2022, 38( 5): 992- 997. DOI: 10.3969/j.issn.1001-5256.2022.05.004.刘秀峰, 张珏, 姚琳, 等. 中晚期肝细胞癌靶向联合免疫治疗进展[J]. 临床肝胆病杂志, 2022, 38( 5): 992- 997. DOI: 10.3969/j.issn.1001-5256.2022.05.004. [6] YANG SS. Evaluation of Clinical Significance of ANKRD22 in Colorectal Cancer[D]. Hangzhou: Zhejiang University, 2019. DOI: 10.27461/d.cnki.gzjdx.2019.001668.杨赛赛. ANKRD22在结直肠癌细胞中表达意义的研究[D]. 杭州: 浙江大学, 2019. DOI: 10.27461/d.cnki.gzjdx.2019.001668 [7] UTSUMI T, HOSOKAWA T, SHICHITA M, et al. ANKRD22 is an N-myristoylated hairpin-like monotopic membrane protein specifically localized to lipid droplets[J]. Sci Rep, 2021, 11( 1): 19233. DOI: 10.1038/s41598-021-98486-8. [8] WANG R, WU YH, ZHU Y, et al. ANKRD22 is a novel therapeutic target for gastric mucosal injury[J]. Biomed Pharmacother, 2022, 147: 112649. DOI: 10.1016/j.biopha.2022.112649. [9] ZHOU HN, LI YM. Conversion therapy for hepatocellular carcinoma[J/OL]. Chin J Hepatic Surg Electron Ed, 2022, 11( 6): 542- 547. DOI: 10.3877/cma.j.issn.2095-3232.2022.06.002.周辉年, 李玉民. 肝癌转化治疗[J/OL]. 中华肝脏外科手术学电子杂志, 2022, 11( 6): 542- 547. DOI: 10.3877/cma.j.issn.2095-3232.2022.06.002. [10] LU SL, YAO JN, YUAN GD, et al. Current status and prospect of clinical and basic research on conversion therapy for hepatocellular carcinoma[J]. Chin J Exp Surg, 2022, 39( 10): 1837- 1843. DOI: 10.3760/cma.j.cn421213-20220210-01043.陆世鎏, 姚建妮, 袁观斗, 等. 肝癌转化治疗临床与基础研究现状与展望[J]. 中华实验外科杂志, 2022, 39( 10): 1837- 1843. DOI: 10.3760/cma.j.cn421213-20220210-01043. [11] PARRA RG, ESPADA R, VERSTRAETE N, et al. Structural and energetic characterization of the ankyrin repeat protein family[J]. PLoS Comput Biol, 2015, 11( 12): e1004659. DOI: 10.1371/journal.pcbi.1004659. [12] LIU X, ZHAO JL, WU Q, et al. ANKRD22 promotes glioma proliferation, migration, invasion, and epithelial-mesenchymal transition by upregulating E2F1-mediated MELK expression[J]. J Neuropathol Exp Neurol, 2023, 82( 7): 631- 640. DOI: 10.1093/jnen/nlad034. [13] WU YG, LIU HX, GONG YF, et al. ANKRD22 enhances breast cancer cell malignancy by activating the Wnt/β-catenin pathway via modulating NuSAP1 expression[J]. Bosn J Basic Med Sci, 2021, 21( 3): 294- 304. DOI: 10.17305/bjbms.2020.4701. [14] YIN J, FU WF, DAI L, et al. ANKRD22 promotes progression of non-small cell lung cancer through transcriptional up-regulation of E2F1[J]. Sci Rep, 2017, 7( 1): 4430. DOI: 10.1038/s41598-017-04818-y. [15] WU YG, CHEN WX, ZHANG B, et al. ANKRD22 knockdown suppresses papillary thyroid cell carcinoma growth and migration and modulates the Wnt/β-catenin signaling pathway[J]. Tissue Cell, 2023, 84: 102193. DOI: 10.1016/j.tice.2023.102193. [16] PAN TH, LIU JW, XU S, et al. ANKRD22, a novel tumor microenvironment-induced mitochondrial protein promotes metabolic reprogramming of colorectal cancer cells[J]. Theranostics, 2020, 10( 2): 516- 536. DOI: 10.7150/thno.37472. [17] CHEN HH, YANG KQ, PANG LX, et al. ANKRD22 is a potential novel target for reversing the immunosuppressive effects of PMN-MDSCs in ovarian cancer[J]. J Immunother Cancer, 2023, 11( 2): e005527. DOI: 10.1136/jitc-2022-005527. [18] QIU YQ, YANG SS, PAN TH, et al. ANKRD22 is involved in the progression of prostate cancer[J]. Oncol Lett, 2019, 18( 4): 4106- 4113. DOI: 10.3892/ol.2019.10738. [19] TIAN LY, SMIT DJ, JÜCKER M. The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism[J]. Int J Mol Sci, 2023, 24( 3): 2652. DOI: 10.3390/ijms24032652. [20] LI YH, YIN YL, HE Y, et al. SOS1 regulates HCC cell epithelial-mesenchymal transition via the PI3K/AKT/mTOR pathway[J]. Biochem Biophys Res Commun, 2022, 637: 161- 169. DOI: 10.1016/j.bbrc.2022.11.015. [21] CHEN JX, CHEN JD, HUANG JX, et al. HIF-2α upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway[J]. Aging(Albany NY), 2019, 11( 23): 10839- 10860. DOI: 10.18632/aging.102488. -

 下载:
下载:

 本文二维码
本文二维码
计量
- 文章访问数: 1641
- HTML全文浏览量: 372
- PDF下载量: 101
- 被引次数: 0

