
 PDF下载 ( 1969 KB)
PDF下载 ( 1969 KB)

-
摘要: 胆汁淤积性肝病(CLD)是指各种病因引起的胆汁代谢异常、流出受阻、胆管损伤等肝脏疾病,主要病因包括:药物、毒物、免疫、遗传、梗阻、感染、肿瘤等。胆汁淤积是CLD共有的病理改变,而不同病因淤胆的部位、组织病理及超微结构等改变,具有相对特异性。依据病因,重点阐述自身免疫性胆管炎、遗传代谢性肝病、大胆管病变的病理学特征,引申鉴别其他CLD,以期提高对CLD病理学的认识,助力精准诊疗。Abstract: Cholestatic liver disease (CLD) is a group of liver diseases caused by various reasons, such as abnormal bile metabolism, blocked outflow, and bile duct injury, and the major causes of CLD include drugs, poisons, immunity, genetics, obstruction, infection, and tumor. Cholestasis is a common pathological change in CLD; however, the site, histopathology, and ultrastructure of cholestasis due to different etiologies are relatively specific. According to the etiology, this article elaborates on the pathological characteristics of CLD such as autoimmune cholangitis, inherited metabolic liver disease, and large bile duct disease and introduces the differential diagnosis of other types of CLD, in order to improve the understanding of CLD pathology and facilitate accurate diagnosis and treatment.
-
Key words:
- Cholestasi /
- Biopsy /
- Pathology /
- Diagnosis
-
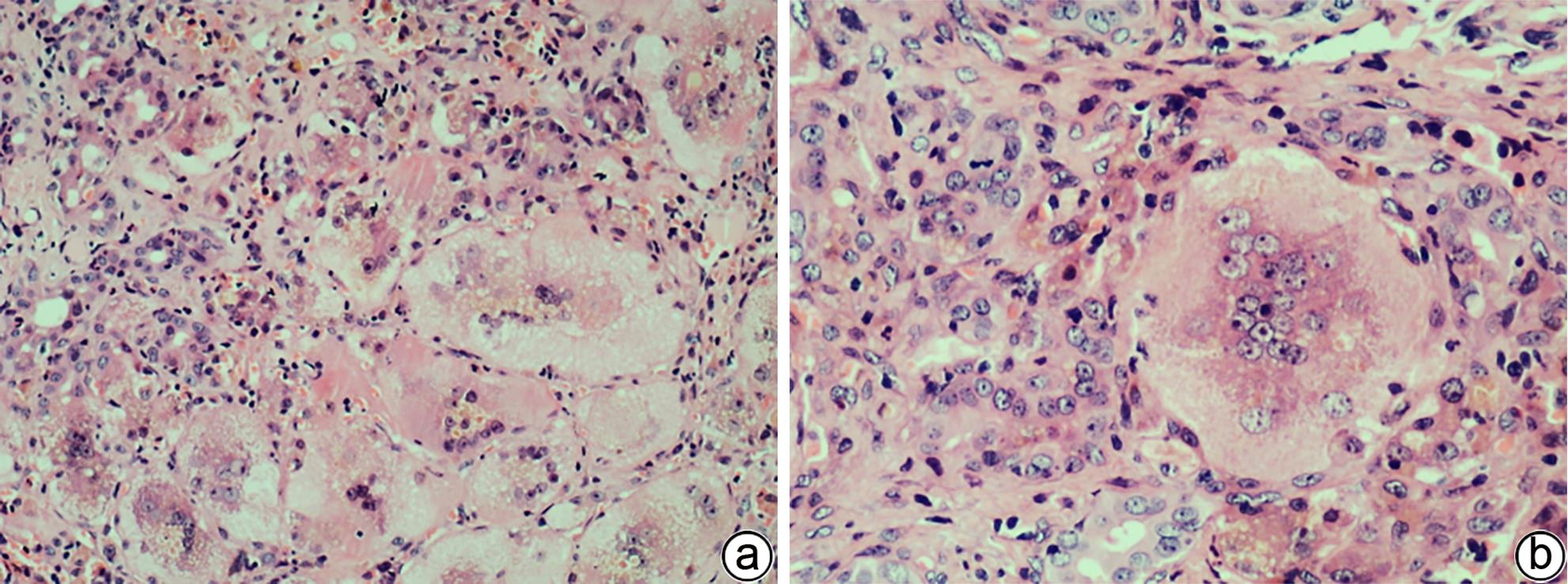
注: a,×100;b,×200。较多巨细胞变肝细胞,胞体巨大,多核,胞质内可见淤胆性色素颗粒,汇管区小胆管损伤及轻微扩张,间质可见中性粒细胞等炎性细胞浸润。
图 1 巨细胞变肝细胞(HE染色)
Figure 1. Hepatocyte with giant-cell transformation(HE staining)

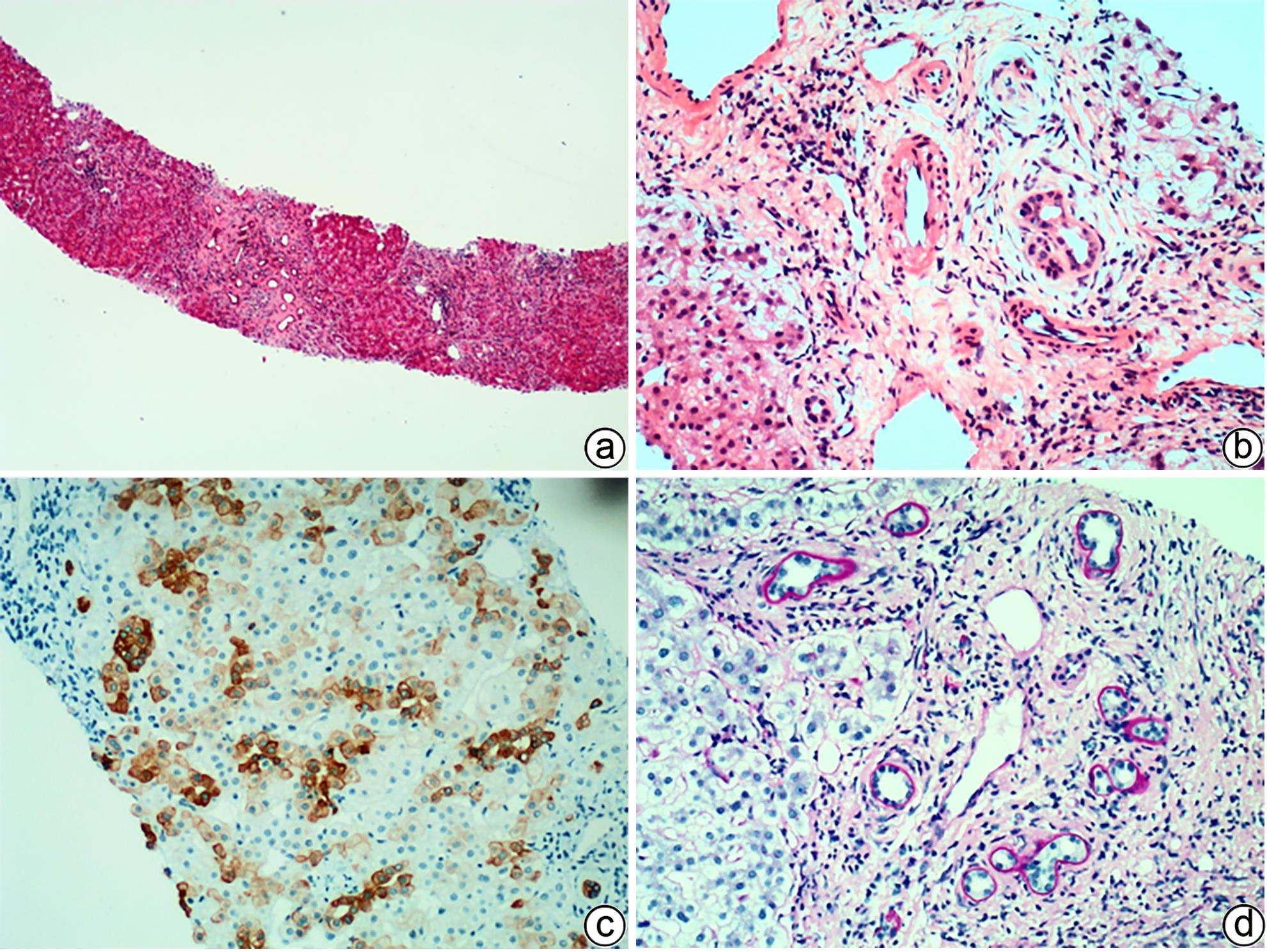
注: a,小胆管损伤伴淋巴细胞、浆细胞包围浸润及上皮样细胞肉芽肿,偶见多核巨细胞(HE染色,×100);b,旺炽型胆管炎,胆管上皮细胞嗜酸性化,核固缩,排列不整齐,管壁及管周较多淋巴细胞、浆细胞聚集浸润,散在少许嗜酸性粒细胞(HE染色,×200);c,CK7标记未见小胆管缺失,可见轻微细胆管反应(免疫组化,×100)。
图 2 PBC典型的病理学特征
Figure 2. Typical pathological features of PBC
注: a,肝硬化,汇管区轻度炎症,小胆管轻度扩张,管周纤维化(HE染色,×20);b,汇管区轻度炎症,小胆管基底膜增厚,周围“洋葱皮样”纤维化,小动脉扩大,管壁轻微增厚,门静脉分支扩张及管周纤维化,汇管区周围肝细胞气球样变性(HE染色,×100);c,免疫组化CK7显示肝细胞胆管化(IHC染色,×100);d,小胆管基底膜增厚(D-PAS染色,×100)。
图 3 PSC典型的病理学特征
Figure 3. Typical histopathological features of PSC
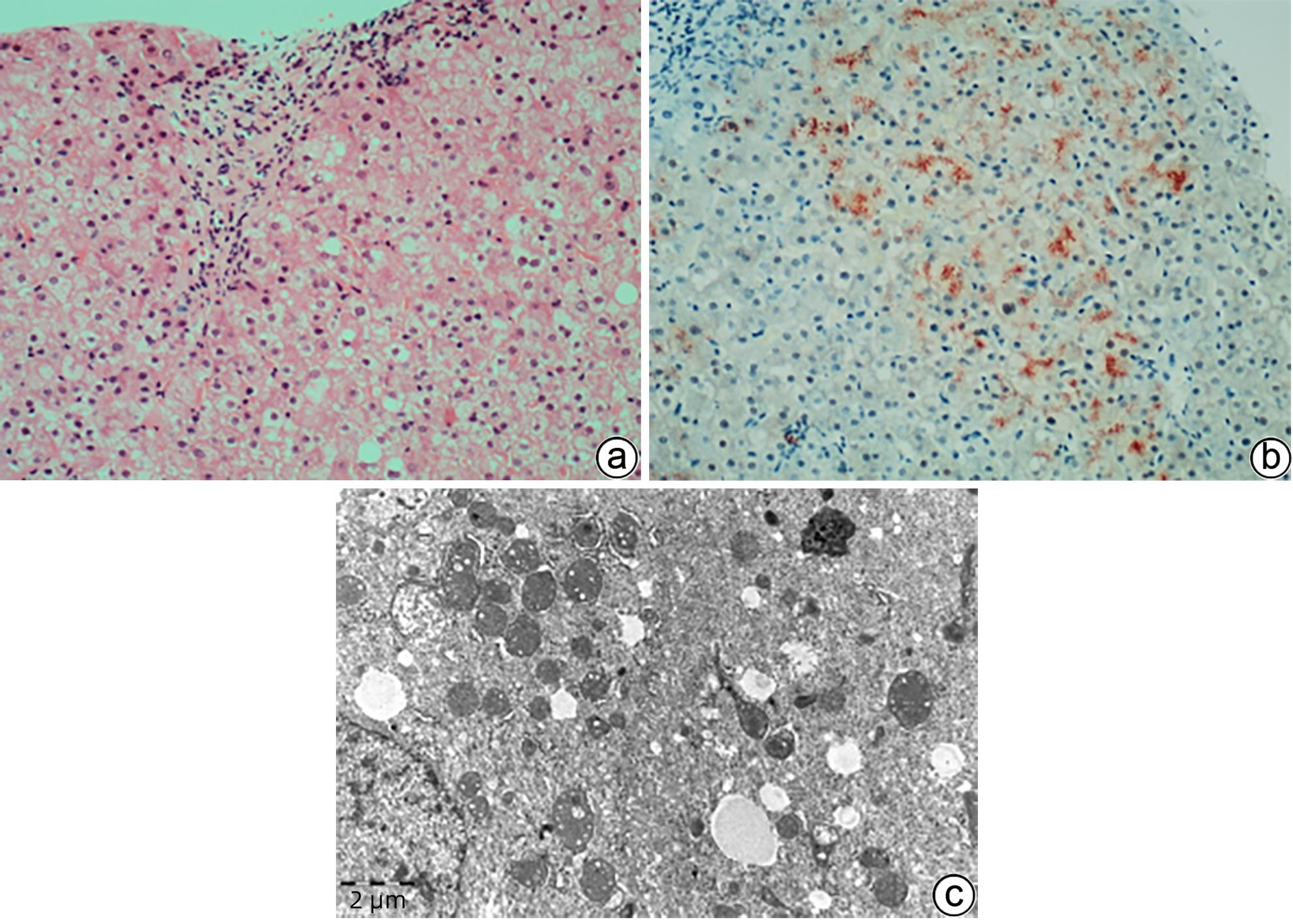
注: a,肝细胞水肿、脂肪变性、气球样变性,部分胞质呈淡红色颗粒状,腺泡1区少许糖原核肝细胞。汇管区轻度炎症(HE染色,×100);b,肝细胞胞质内较多红色铜颗粒沉积(罗丹宁铜染色,×100);c,肝细胞胞质内线粒体形态不规则,内外膜分类,可见内嵴扩张空泡及圆形高电子致密颗粒。肝细胞胞质内也可见脂滴、含高电子致密度的淤胆性色素颗粒沉积(TEM,×12 000)。
图 4 肝豆状核变性的组织病理学及超微结构特征
Figure 4. Histopathological and ultrastructural characteristics of WD
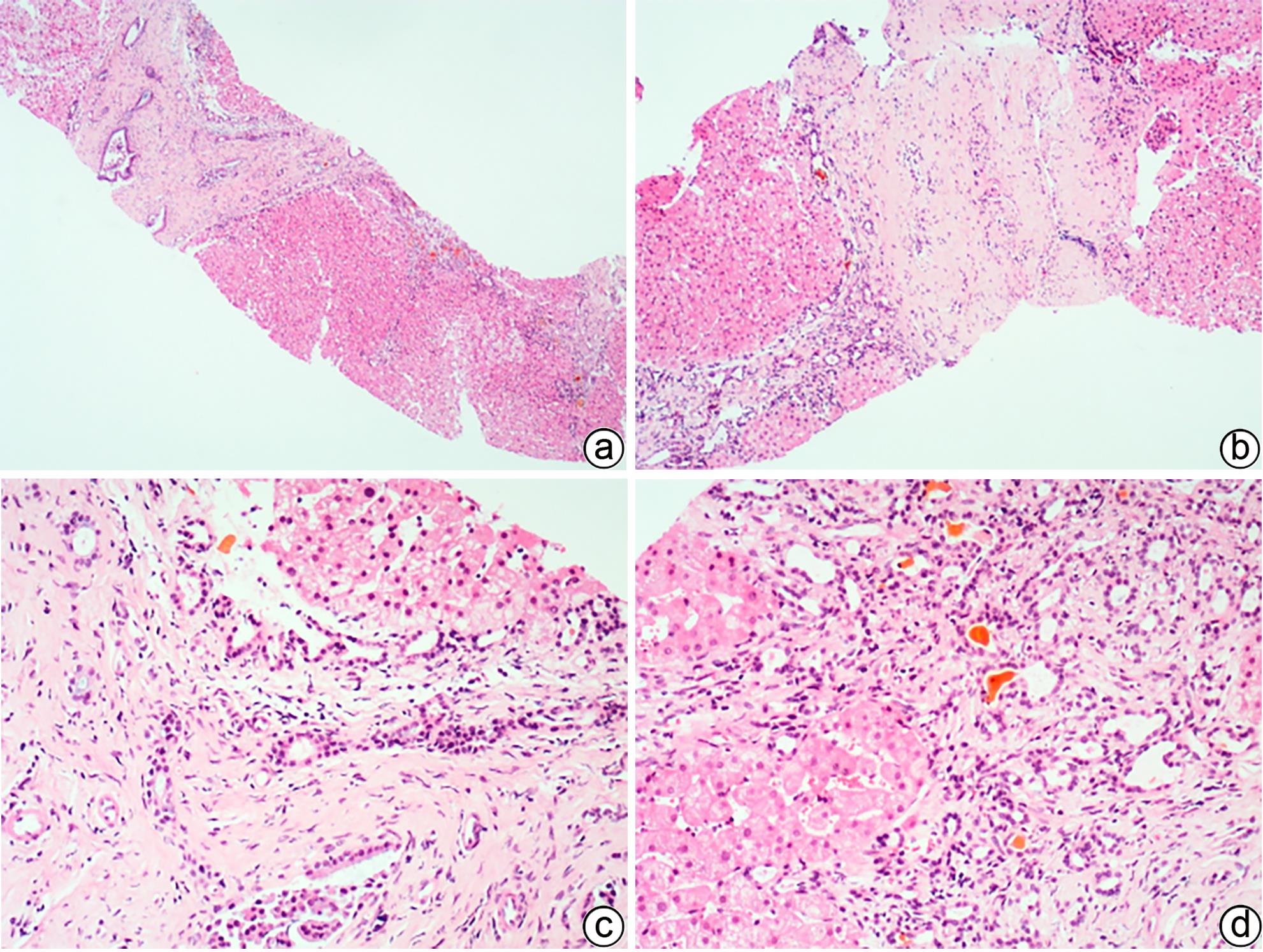
注: a,CHF“地图样”纤维化,胆管板畸形,胆汁淤积(HE染色,×40);b,汇管区胆管板畸形,门静脉狭窄、闭塞,肝细胞轻度水肿(HE染色,×100);c,畸形的胆管板内陷于汇管区纤维组织中,门静脉分支狭窄、闭塞(HE染色,×200);d,胆管板畸形,形态不规则,管腔扩张,可见胆栓,间质少许淋巴细胞、浆细胞浸润(HE染色,×200)。
图 5 CHF典型的组织病理学特征
Figure 5. Typical histopathological features of CHF

注: a,肝细胞及毛细胆管内两种颜色的淤胆颗粒及胆栓,一种“巧克力”色,另一种淡黄色(HE染色,×100);b,偏振光显微镜可见红色双折光中的“Maltese十字”或星状暗区(HE染色,偏振光,×200);c,毛细胆管内可见原卟啉结晶(箭头),呈丝状弧形排列,毛细胆管腔面微绒毛减少,紧密连接延长(TEM,×15 000);d,Kupffer细胞胞质溶酶体内充满丝状原卟啉结晶(箭头),窦周间隙可见胶原纤维束沉积(TEM,×15 000)。
图 6 EPP的组织病理学及超微结构特征
Figure 6. Histopathological and ultrastructural characteristics of EPP
-
[1] JOHNSON CA, GISSEN P, SERGI C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes[J]. J Med Genet, 2003, 40( 5): 311- 319. DOI: 10.1136/jmg.40.5.311. [2] ESTRADAS J, PASCUAL-RAMOS V, MARTÍNEZ B, et al. Autoimmune hepatitis with giant-cell transformation[J]. Ann Hepatol, 2009, 8( 1): 68- 70. [3] SARCOGNATO S, SACCHI D, GRILLO F, et al. Autoimmune biliary diseases: Primary biliary cholangitis and primary sclerosing cholangitis[J]. Pathologica, 2021, 113( 3): 170- 184. DOI: 10.32074/1591-951X-245. [4] ZEN Y, HUBSCHER SG, NAKANUMA Y. Bile duct diseases. BurtAD, FerrellLD, HübscherSG, eds. MacSween’s pathology of the liver[M]. 7th ed. Philadelphia, PA: Elsevier, 2018: 515. [5] TAKAHASHI T, MIURA T, NAKAMURA J, et al. Plasma cells and the chronic nonsuppurative destructive cholangitis of primary biliary cirrhosis[J]. Hepatology, 2012, 55( 3): 846- 855. DOI: 10.1002/hep.24757. [6] CAREY EJ, ALI AH, LINDOR KD. Primary biliary cirrhosis[J]. Lancet, 2015, 386: 1565- 1575. DOI: 10.1016/S0140-6736(15)00154-3. [7] KARLSEN TH, FOLSERAAS T, THORBURN D, et al. Primary sclerosing cholangitis-a comprehensive review[J]. J Hepatol, 2017, 67( 6): 1298- 1323. DOI: 10.1016/j.jhep.2017.07.022. [8] HIRSCHFIELD GM, KARLSEN TH, LINDOR KD, et al. Primary sclerosing cholangitis[J]. Lancet, 2013, 382( 9904): 1587- 1599. DOI: 10.1016/S0140-6736(13)60096-3. [9] PORTMANN B, ZEN Y. Inflammatory disease of the bile ducts-cholangiopathies: Liver biopsy challenge and clinicopathological correlation[J]. Histopathology, 2012, 60( 2): 236- 248. DOI: 10.1111/j.1365-2559.2011.03853.x. [10] COLLING R, VERRILL C, FRYER E, et al. Bile duct basement membrane thickening in primary sclerosing cholangitis[J]. Histopathology, 2016, 68( 6): 819- 824. DOI: 10.1111/his.12857. [11] FIEL MI, SIMA HR, AZARIAN A, et al. A morphometric study of the hepatic arterioles in end-stage primary sclerosing cholangitis[J]. Virchows Arch, 2015, 466( 2): 143- 149. DOI: 10.1007/s00428-014-1680-9. [12] CARRASCO-AVINO G, SCHIANO TD, WARD SC, et al. Primary sclerosing cholangitis: Detailed histologic assessment and integration using bioinformatics highlights arterial fibrointimal hyperplasia as a novel feature[J]. Am J Clin Pathol, 2015, 143( 4): 505- 513. DOI: 10.1309/AJCPVKFVIPRBXQR2. [13] NAKAZAWA T, NAITOH I, HAYASHI K, et al. Diagnostic criteria for IgG4-related sclerosing cholangitis based on cholangiographic classification[J]. J Gastroenterol, 2012, 47( 1): 79- 87. DOI: 10.1007/s00535-011-0465-z. [14] ZHANG JP, HOU XT, YIN ZC, et al. Gilbert syndrome: Clinicopathological and genetic analyses of 29 cases[J]. Chin J Diagn Pathol, 2018, 25( 2): 85- 89. DOI: 10.3969/j.issn.1007-8096.2018.02.002.张继平, 侯晓涛, 尹自长, 等. Gilbert综合征29例临床病理及基因分析[J]. 诊断病理学杂志, 2018, 25( 2): 85- 89. DOI: 10.3969/j.issn.1007-8096.2018.02.002. [15] ATAOLLAHI M, DEHGHANI SM, ANBARDAR MH, et al. Liver histologic changes in children with type 1 of Crigler-Najjar syndrome[J]. Arkh Patol, 2021, 83( 5): 27- 30. DOI: 10.17116/patol20218305127. [16] FATA CR, GILLIS LA, PACHECO MC. Liver fibrosis associated with crigler-najjar syndrome in a compound heterozygote: A case report[J]. Pediatr Dev Pathol, 2017, 20( 6): 522- 525. DOI: 10.1177/1093526617697059. [17] WU ZB. Ultramicro-pathological diagnostics[M]. Shanghai: Shanghai Scientific& Technical Publishers, 2003.武忠弼. 超微病理诊断学[M]. 上海: 上海科学技术出版社, 2003. [18] LI LT, WANG JS. Advances in the study of progressive familial intrahepatic cholestasis[J]. Infect Dis Inf, 2019, 32( 2): 162- 165. DOI: 10.3969/j.issn.1007-8134.2019.02.017.李丽婷, 王建设. 进行性家族性肝内胆汁淤积症研究进展[J]. 传染病信息, 2019, 32( 2): 162- 165. DOI: 10.3969/j.issn.1007-8134.2019.02.017. [19] WENG YH, XIONG QF, LIU DX, et al. Clinical and pathological features of progressive familial intrahepatic cholestasis type 3[J]. J Clin Hepatol, 2022, 38( 1): 154- 159. DOI: 10.3969/j.issn.1001-5256.2022.01.024.翁宇航, 熊清芳, 刘杜先, 等. 进行性家族性肝内胆汁淤积症3型临床病理特征分析[J]. 临床肝胆病杂志, 2022, 38( 1): 154- 159. DOI: 10.3969/j.issn.1001-5256.2022.01.024. [20] QIU YL, GONG JY, FENG JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis[J]. Hepatology, 2017, 65( 5): 1655- 1669. DOI: 10.1002/hep.29020. [21] GOMEZ-OSPINA N, POTTER CJ, XIAO R, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis[J]. Nat Commun, 2016, 7: 10713. DOI: 10.1038/ncomms10713. [22] HALAWI A, IBRAHIM N, BITAR R. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: A review of literature[J]. Acta Gastroenterol Belg, 2021, 84( 3): 477- 486. DOI: 10.51821/84.3.013. [23] GUINDI M. Wilson disease[J]. Semin Diagn Pathol, 2019, 36( 6): 415- 422. DOI: 10.1053/j.semdp.2019.07.008. [24] FANNI D, GUIDO M, GEROSA C, et al. Liver changes in Wilson’s disease: The full spectrum. A report of 127 biopsies from 43 patients[J]. Eur Rev Med Pharmacol Sci, 2021, 25( 12): 4336- 4344. DOI: 10.26355/eurrev_202106_26142. [25] ZHAO XY, HE ZY, LIU LW, et al. Comparative study of pathological characteristics of 45 patients with primary and secondary hemochromatosis[J]. Infect Dis Inf, 2019, 32( 2): 127- 131. DOI: 10.3969/j.issn.1007-8134.2019.02.007.赵新颜, 何志颖, 刘立伟, 等. 45例原发性与继发性血色病临床病理特点对比研究[J]. 传染病信息, 2019, 32( 2): 127- 131. DOI: 10.3969/j.issn.1007-8134.2019.02.007. [26] MIYAMOTO R, JUN SD, OTA K, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency with no hepatic steatosis: A case report[J]. BMC Pediatr, 2021, 21( 1): 237. DOI: 10.1186/s12887-021-02717-w. [27] ZHANG JP, CHENG YB, ZHOU XJ, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: A clinicopathological analysis of two cases[J]. Chin J Diagn Pathol, 2018, 25( 4): 261- 265. DOI: 10.3969/j.issn.1007-8096.2018.04.006.张继平, 程艳波, 周晓军, 等. Citrin缺陷导致的新生儿肝内胆汁淤积症2例临床病理观察[J]. 诊断病理学杂志, 2018, 25( 4): 261- 265. DOI: 10.3969/j.issn.1007-8096.2018.04.006. [28] FABRIS L, MILANI C, FIOROTTO R, et al. Dysregulation of the Scribble/YAP/β-catenin axis sustains the fibroinflammatory response in a PKHD1-/- mouse model of congenital hepatic fibrosis[J]. FASEB J, 2022, 36( 6): e22364. DOI: 10.1096/fj.202101924R. [29] SAXENA R. Practical hepatic pathology: A diagnostic approach[M]. 2nd Ed. Philadelphia: Elsevier, 2018. [30] DESMET VJ. Congenital diseases of intrahepatic bile ducts: Variations on the theme“ductal plate malformation”[J]. Hepatology, 1992, 16( 4): 1069- 1083. DOI: 10.1002/hep.1840160434. [31] CHEN IY, WHITNEY-MILLER CL, LIAO XY. Congenital hepatic fibrosis and its mimics: A clinicopathologic study of 19 cases at a single institution[J]. Diagn Pathol, 2021, 16( 1): 81. DOI: 10.1186/s13000-021-01142-y. [32] GILBERT MA, BAUER RC, RAJAGOPALAN R, et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification[J]. Hum Mutat, 2019, 40( 12): 2197- 2220. DOI: 10.1002/humu.23879. [33] WU LN, SUN LY, ZHU ZJ, et al. Clinical and histological characteristics of patients with Alagille syndrome[J]. Chin Hepatol, 2023, 28( 3): 351- 354, 363. DOI: 10.14000/j.cnki.issn.1008-1704.2023.03.018.武丽娜, 孙丽莹, 朱志军, 等. Alagille综合征的临床及病理特征分析[J]. 肝脏, 2023, 28( 3): 351- 354, 363. DOI: 10.14000/j.cnki.issn.1008-1704.2023.03.018. [34] CASANOVA-GONZÁLEZ MJ, TRAPERO-MARUGÁN M, JONES EA, et al. Liver disease and erythropoietic protoporphyria: A concise review[J]. World J Gastroenterol, 2010, 16( 36): 4526- 4531. DOI: 10.3748/wjg.v16.i36.4526. [35] ANSTEY AV, HIFT RJ. Liver disease in erythropoietic protoporphyria: Insights and implications for management[J]. Gut, 2007, 56( 7): 1009- 1018. DOI: 10.1136/gut.2006.097576. [36] MACDONALD DM, GERMAIN D, PERROT H. The histopathology and ultrastructure of liver disease in erythropoietic protoporphyria[J]. Br J Dermatol, 1981, 104( 1): 7- 17. DOI: 10.1111/j.1365-2133.1981.tb01705.x. [37] VIJ M, RELA M. Biliary atresia: Pathology, etiology and pathogenesis[J]. Future Sci OA, 2020, 6( 5): FSO466. DOI: 10.2144/fsoa-2019-0153. -

 下载:
下载:
 本文二维码
本文二维码
计量
- 文章访问数: 3012
- HTML全文浏览量: 1119
- PDF下载量: 474
- 被引次数: 0

