PDF下载 ( 1944 KB)
PDF下载 ( 1944 KB)

Research advances in animal models of primary biliary cholangitis
-
摘要: 原发性胆汁性胆管炎(PBC)是由针对肝内中小胆管的自身免疫反应引起的胆汁淤积性肝脏炎症疾病。临床上最具疾病特异性的表现为自身抗线粒体抗体阳性和肝脏组织学上的选择性中小胆管破坏。由于PBC早期临床样本难以获取,构建并优化合适的小鼠模型是研究PBC发病机制的重要手段。了解PBC动物模型的建模原理和在血清学、组织学、细胞学上的疾病特征表现,既有利于加深对PBC疾病的认识,也有助于科学、合理地设计研究方案。Abstract: Primary biliary cholangitis (PBC) is an inflammatory and cholestatic liver disease caused by autoimmune response targeting the small- and medium-sized intrahepatic bile ducts. The most specific manifestations of this diseases in clinical practice were positive anti-mitochondrial antibody and selective destruction of small- and medium-sized bile ducts based on liver histology. Since it is difficult to obtain the clinical samples of early-stage PBC, the construction and optimization of mouse models is an important method to investigate the pathogenesis of PBC. An understanding of the modeling principles of PBC animal models and disease features in serology, histology, and cytology not only helps to improve the awareness of PBC, but also helps to design scientific and rational research protocols.
-
Key words:
- Autoimmune Diseases /
- Liver Cirrhosis, Biliary /
- Disease Models, Animal
-
全球有超过2.4亿慢性HBV感染者,若不及时进行有效、规范的治疗,15%~40%的患者会进展为肝硬化,最终导致肝衰竭和肝细胞癌(HCC)[1]。虽然乙型肝炎疫苗的接种使HBV感染的患病率逐年下降,但多数亚洲地区仍归为中至高流行区。我国HBsAg流行率为5%~6%,但因为人口基数大,所以仍存在许多的慢性HBV感染者,其中需要治疗的慢性乙型肝炎(CHB)患者约有2000万~3000万[2-3];因此慢性HBV感染依然是我国的重大公共卫生问题。当前用于抗病毒治疗的核苷(酸)类似物(nucleos(t)ide analogues, NAs)虽可有效抑制病毒复制,延缓疾病进展,但对肝细胞核内的共价闭合环状DNA(covalently closed circular DNA,cccDNA)均无明确作用,使得病毒无法完全清除。cccDNA作为HBV复制的原始模板,检测肝内cccDNA水平是评价抗病毒疗效及停止治疗的重要指标[4-5],由于肝活检为侵入性操作,cccDNA在肝组织内分布不均,松弛环状双链DNA(relaxed circularDNA,rcDNA)的存在影响cccDNA含量等因素导致cccDNA检测难以在临床广泛开展[6-7]。因此需要寻找能反映肝内cccDNA活性且方便操作的临床替代指标。近年来HBV RNA作为新的血清学标志物被广泛提出,因为其只能来自肝内cccDNA, 所以能更好的反映HBV转录活性,本研究主要探讨血清HBV RNA在HBeAg阳性CHB患者不同时期的表达水平及检测价值。
1. 资料与方法
1.1 研究对象
收集2019年8月—2020年12月在杭州市西溪医院肝病科门诊及住院部诊治的CHB患者,纳入标准:(1)诊断符合《慢性乙型肝炎防治指南(2019年版)》[8];(2)准备接受或已接受NAs抗病毒治疗。排除标准:(1)合并HAV、HCV、巨细胞病毒等其他嗜肝病毒感染;(2)合并HIV感染;(3)HCC患者;(4)代谢性肝病、自身免疫性肝病及近期使用肝损伤性药物者;(5)合并其他系统恶性肿瘤或严重疾病者;(6)研究者认为不适合入组的其他情况。
1.2 实验室检测
本研究使用的HBV RNA定量检测试剂盒由湖南圣湘科技有限公司提供,检测原理通过逆转录HBV核酸中的pgRNA(经DNA酶消化),利用针对HBV pgRNA序列设计的一组特异性引物与荧光探针,配以PCR反应液,在荧光定量PCR仪上,应用一步法RT实时荧光定量PCR检测技术,通过荧光信号的变化实现HBV pgRNA的定量检测,HBV RNA检测下限为50拷贝/mL。应用化学发光免疫分析法在美国雅培Alinity i全自动化学发光免疫分析仪上行HBV血清学标志物检测;用HBV DNA定量检测试剂盒,在ABI7500荧光定量PCR仪上行HBV DNA检测;HBV DNA检测下限为30 IU/mL;用Beckman Coulter AU5831全自动生化分析仪检测ALT(正常范围9~50 U/L)、AST(正常范围15~40 U/L)。
1.3 伦理学审查
本研究经杭州市西溪医院伦理委员会批准, 批号:2019年(科)伦审第21号,所有患者均签署知情同意书。
1.4 统计学方法
采用SPSS 25.0进行统计学处理,正态分布的计量资料用x ±s表示,2组间比较采用独立样本t检验;非正态分布的计量资料用M(P25~P75)表示,2组间比较采用Mann-Whitney U检验;计数资料组间比较采用χ2检验;采用Pearson或Spearman相关分析描述两变量间的相关性。P<0.05为差异有统计学意义。
2. 结果
2.1 一般资料
本研究共纳入61例CHB患者,平均年龄(39.95±9.88)岁。按HBeAg及HBV DNA状态分为3组:HBeAg阳性CHB[HBeAg(+)、HBV DNA(+)]未治患者,HBeAg血清学转换前[HBeAg(+)、HBV DNA(-)]经治患者,HBeAg血清学转换后[HBeAg(-)、HBV DNA(-)]经治患者(表 1)。
表 1 纳入患者分组情况及基线资料组别 例数 男 女 年龄(岁) HBeAg(+)、HBV DNA(+) 22 13 9 35.36±7.55 HBeAg(+)、HBV DNA(-) 17 11 6 39.35±7.98 HBeAg(-)、HBV DNA(-) 22 13 9 45.00±11.16 2.2 不同时期血清HBV RNA的表达水平
HBeAg阳性CHB未治患者血清HBV RNA阳性率100%(22/22),HBV RNA载量最大值为9 log10拷贝/mL,平均为7 log10拷贝/mL;HBeAg血清学转换前经治患者血清HBV RNA阳性率88.2%(15/17),HBV RNA载量最大值为5 log10拷贝/mL,平均4 log10拷贝/mL;HBeAg血清学转换后经治患者血清HBV RNA阳性率22.7%(6/22),HBV RNA载量最大值为4 log10拷贝/mL。
2.3 经治HBeAg阳性组与HBeAg阴性组血清学指标比较
经治HBeAg阳性组的HBV RNA阳性率显著高于HBeAg阴性组,差异有统计学意义(P<0.001), 2组间HBV RNA、HBsAg水平比较,差异均有统计学意义(P值均<0.05)(表 2)。
表 2 经治患者HBeAg阳性组与HBeAg阴性组临床资料比较项目 HBeAg阳性组(n=17) HBeAg阴性组(n=22) 统计值 P值 男/女(例) 11/6 13/9 0.753 年龄(岁) 39.35±7.98 45.00±11.16 t=-1.77 0.086 HBV RNA阳性[例(%)] 15(88.2) 6(27.3) <0.001 HBV RNA(log10拷贝/mL) 4.05(3.01~5.18) 1.40(1.40~1.83) Z=-4.44 <0.001 HBsAg(log10IU/mL) 3.24(2.86~3.50) 2.76(2.07~3.35) Z=-2.41 0.016 ALT(U/L) 22.24±10.83 25.09±11.76 t=-0.78 0.442 AST(U/L) 23.00±4.48 23.9±14.23 t=-0.65 0.521 2.4 HBV DNA阳性组与HBV DNA阴性组血清学指标比较
CHB患者NAs治疗实现HBV DNA阴转时,肝功能复常,ALT、AST水平降低,HBV RNA、HBsAg水平也均有所降低(P值均<0.001)(表 3)。对于治疗后HBV DNA阴转的CHB患者,Spearman相关分析示,血清HBV RNA与血清HBsAg无相关性(P=0.091)。
表 3 HBV DNA阳性组与HBV DNA阴性组临床资料比较项目 HBV DNA阳性组(n=22) HBV DNA阴性组(n=39) 统计值 P值 男/女(例) 13/9 24/15 χ2=0.035 0.851 年龄(岁) 35.36±7.55 42.54±10.18 t=-2.88 0.005 HBV RNA(log10拷贝/mL) 7.62(6.51~8.55) 1.94(1.40~4.04) Z=-6.16 <0.001 HBsAg(log10IU/mL) 3.94(3.26~4.23) 2.90(2.61~3.44) Z=-4.07 <0.001 ALT(U/L) 249.50(114.25~664.50) 20.00(16.00~31.00) Z=-6.45 <0.001 AST(U/L) 106.00(63.75~418.75) 23.00(21.00~27.00) Z=-6.45 <0.001 HBV DNA(log10IU/ml) 7.21±1.23 - 2.5 不同时期HBV RNA与HBsAg、HBV DNA的相关性
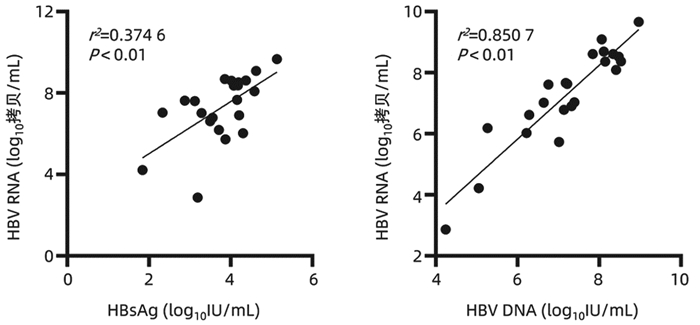
在HBeAg阳性CHB未治患者中,HBV RNA与HBsAg(r=0.612,P<0.01)、HBV DNA(r=0.922, P<0.01)均具有较强的相关性(图 1);在HBeAg血清学转换前和HBeAg血清学转换后的经治患者中,HBV RNA与HBsAg无相关性(P>0.05);3组患者HBV RNA与年龄、ALT、AST均无相关性(P值均>0.05)。
3. 讨论
CHB是导致肝硬化、肝衰竭、HCC的主要危险因素,每年约有80万人死于HBV感染相关性疾病[9],因此如何有效的管理CHB患者仍是当前临床工作的热点和难点。随着对HBV RNA的研究深入,已知HBV感染患者血清中的HBV RNA就是前基因组RNA(pgRNA),即3.5 kb mRNA;pgRNA是HBV复制的中间产物,利用cccDNA作为模板在病毒核衣壳内转录,最后释放完整的子代病毒,因此血清HBV RNA与HBsAg不同,其只能来自于肝内cccDNA;而NAs是通过取代HBV复制过程中聚合酶区的核苷来抑制病毒复制,并不能影响cccDNA转录生成mRNA,由此可以推断HBV RNA与肝内cccDNA相关,检测外周血HBV RNA水平可以反映肝内cccDNA的转录活性。因此相比于HBsAg、HBV DNA等传统的血清学指标,血清HBV RNA水平可以更好的反映肝内病毒复制水平,其能作为CHB患者管理的新型标志物[10-11]。
HBeAg阳性CHB患者在接受抗病毒药物治疗后会逐步实现HBV DNA阴转、HBeAg血清学转换等目标,本研究通过观察HBeAg阳性的CHB患者不同时期的HBV RNA表达水平,发现未治疗的22例CHB患者血清中均能检测出较高的血清HBV RNA水平;39例治疗后HBV DNA阴转的CHB患者中仍有21例可以检测出HBV RNA,这表明在评估病毒复制方面HBV RNA比HBV DNA更灵敏,即使是在HBV DNA持续低于检测下限且发生HBeAg血清学转换也有部分患者血清HBV RNA阳性,意味HBeAg的消失只能表示病毒复制减弱,肝内cccDNA仍可能存在低水平的转录。
对HBeAg阳性CHB患者而言,发生HBeAg血清学转换是比较满意的终点,是发生HBsAg阴转的基本条件。本文通过比较治疗后HBV DNA阴转的HBeAg阳性组与HBeAg阴性组,发现HBeAg阳性组HBV RNA阳性率显著高于HBeAg阴性组(88.2% vs 27.3%),两组间HBV RNA、HBsAg水平差异也具有统计学意义,因HBV RNA只能来自于肝内cccDNA, 提示HBeAg阳性CHB患者肝内cccDNA转录活性更高。因此临床上对HBeAg阳性的CHB患者要努力实现HBeAg血清学转换。
通过分析HBV DNA阳性组与HBV DNA阴性组患者,发现接受NAs治疗后HBV DNA、HBV RNA均会发生下降,这是因为NAs类药物抑制pgRNA的逆转录,致使DNA合成受阻,而长时间接受NAs治疗使rcDNA的形成受到抑制,影响cccDNA池的回补,被感染的肝细胞数量减少,进一步导致HBV RNA的生成也减少;另外接受NAs治疗的CHB人群在病毒清除的过程中可能会促进机体免疫应答的能力从而减少cccDNA池。但是在临床上发现即使CHB患者经过长期NAs抗病毒治疗,能实现完全治愈的患者极为罕见,可能是只需要极少部分逆转录酶存在活性就能对cccDNA池进行补充,因此对大部分CHB患者而言需要长期口服NAs抗病毒治疗。
本研究发现,未接受治疗的CHB患者HBV RNA与HBsAg(r=0.612,P<0.01)、HBV DNA(r=0.922, P<0.01)均具有较强的正相关,这种血清学间良好的相关性有助于基层医院或者医疗资源匮乏地区,使用HBsAg定量水平反映HBeAg阳性CHB患者体内病毒复制水平,而另外两组HBV DNA阴性的CHB患者HBV RNA与HBsAg无相关性,这与Mak等[12]的研究结果一致,随着治疗时间的延长,HBV RNA与HBsAg相关性逐渐下降,从一定程度反映了治疗后的HBsAg水平不能准确反映肝内cccDNA转录活性。
综上所述,对未接受抗病毒治疗的CHB患者,血清HBV RNA与其他血清学标志物有一定的相关性,对NAs治疗后HBV DNA阴转的患者有望使用HBV RNA继续监测肝内病毒复制活性,为CHB患者长期抗病毒治疗提供依据。但本研究样本量较少,相关结论有待进一步证实,后续可以在未治疗组继续纳入合适患者并进行前瞻性队列研究,观察HBeAg阳性CHB患者在NAs治疗过程中HBV RNA的动态变化。
-
表 1 不同PBC模型特点比较
PBC模型 造模原理 优点 缺点 并发症 主要致病细胞 模型选择建议 dnTGFβRⅡ T淋巴细胞中的TGFβ信号通路被阻断 模拟了PBC早期的关键血清学、病理学特征 门静脉区未见嗜酸性粒细胞浸润和肉芽肿 炎症性肠病 CD8+ T淋巴细胞(PBC) 适于探究PBC早期发病过程 未见循环IgM的持续升高,而是出现IgA水平升高 CD4+ T淋巴细胞(结肠炎) ARE-Del-/- 慢性、持续性表达性IFNγ 明显的性别倾向 检测不到ALP
肝纤维化程度较轻系统性红斑狼疮 CD4+ T淋巴细胞 适于探究PBC的性别差异 NOD.c3c4 NOD模型鼠获得胰岛素依赖性糖尿病抗性 部分小鼠可见嗜酸性粒细胞浸润、上皮样肉芽肿样病变和早期肝纤维化 自身免疫的初始攻击位置与临床不同 肝脏胆道多囊病 CD4+ T淋巴细胞和CD8+ T淋巴细胞单独均可致病 - AMA阳性率较低 检测不到ALP 血清中IgM和IgA均升高 Scurfy Treg细胞完全缺陷 - 全身性的自身免疫性疾病 全身性的自身免疫病 CD8+ T淋巴细胞 - 寿命只有4周左右 IL-2Rα-/- Treg细胞减少 - 没有肉芽肿和嗜酸性粒细胞浸润 严重贫血 CD8+ T淋巴细胞(PBC) - 淋巴组织增生性自身免疫病 CD4+ T淋巴细胞(结肠炎) 溃疡性结肠炎 IL-2Rα-/-
IL12-p40-/-在IL-2Rα-/-模型的基础上敲除IL-12p40 抑制了IL-2Rα-/-模型的结肠炎 - 干燥综合征 CD8+ T淋巴细胞 适于探究PBC重度炎症及向纤维化发展的发病过程 加重了肝门静脉炎症和胆管损伤,并促进其向肝纤维化发展 贫血和髓外造血 Ae2a, b-/- 破坏Cl-/HCO3-跨质膜交换,干扰细胞内pH调控 - 个体差异大 - - - 2OA-BSA免疫 用能被AMA识别的半抗原进行长期诱导 可见上皮样肉芽肿样病变 检测不到胆汁淤积酶 - CD8+ T淋巴细胞 适于探究环境因素引起的PBC早期发病过程 不能进展为肝脏纤维化 2OA-poly Ⅰ∶C联合免疫 在2OA-BSA免疫模型的基础上加以病毒RNA模拟物诱导 促进了2OA免疫小鼠的PBC进展 - - - 适于探究环境因素引起的PBC发病过程 可见嗜酸性粒细胞浸润 部分小鼠出现肝纤维化 注: “-”表示特征不显著或鲜有报道。  下载: 导出CSV
下载: 导出CSV
-
[1] SCHEUER PJ. Ludwig Symposium on biliary disorders--part Ⅱ. Pathologic features and evolution of primary biliary cirrhosis and primary sclerosing cholangitis[J]. Mayo Clin Proc, 1998, 73(2): 179-183. DOI: 10.4065/73.2.179. [2] VLEGGAAR FP, van BUUREN HR. No prognostic significance of antimitochondrial antibody profile testing in primary biliary cirrhosis[J]. Hepatogastroenterology, 2004, 51(58): 937-940. [3] GORELIK L, FLAVELL RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease[J]. Immunity, 2000, 12(2): 171-181. DOI: 10.1016/s1074-7613(00)80170-3. [4] HUANG MX, YANG SY, LUO PY, et al. Gut microbiota contributes to sexual dimorphism in murine autoimmune cholangitis[J]. J Leukoc Biol, 2021. DOI: 10.1002/JLB.3MA0321-037R.[Online abead of print] [5] NAKAMURA A, YAMAZAKI K, SUZUKI K, et al. Increased portal tract infiltration of mast cells and eosinophils in primary biliary cirrhosis[J]. Am J Gastroenterol, 1997, 92(12): 2245-2249. [6] HODGE DL, BERTHET C, COPPOLA V, et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice[J]. J Autoimmun, 2014, 53: 33-45. DOI: 10.1016/j.jaut.2014.02.003. [7] BAE HR, LEUNG PS, TSUNEYAMA K, et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance[J]. Hepatology, 2016, 64(4): 1189-1201. DOI: 10.1002/hep.28641. [8] IRIE J, WU Y, WICKER LS, et al. NOD. c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis[J]. J Exp Med, 2006, 203(5): 1209-1219. DOI: 10.1084/jem.20051911. [9] ZHANG W, SHARMA R, JU ST, et al. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis[J]. Hepatology, 2009, 49(2): 545-552. DOI: 10.1002/hep.22651. [10] GODFREY VL, WILKINSON JE, RUSSELL LB. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse[J]. Am J Pathol, 1991, 138(6): 1379-1387. [11] NELSON BH. IL-2, regulatory T cells, and tolerance[J]. J Immunol, 2004, 172(7): 3983-3988. DOI: 10.4049/jimmunol.172.7.3983. [12] WAKABAYASHI K, LIAN ZX, MORITOKI Y, et al. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis[J]. Hepatology, 2006, 44(5): 1240-1249. DOI: 10.1002/hep.21385. [13] HSU W, ZHANG W, TSUNEYAMA K, et al. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin-2Ralpha(-/-) mice[J]. Hepatology, 2009, 49(1): 133-140. DOI: 10.1002/hep.22591. [14] ZHU J, YAMANE H, PAUL WE. Differentiation of effector CD4 T cell populations (*)[J]. Annu Rev Immunol, 2010, 28: 445-489. DOI: 10.1146/annurev-immunol-030409-101212. [15] YAO Y, YANG W, YANG YQ, et al. Distinct from its canonical effects, deletion of IL-12p40 induces cholangitis and fibrosis in interleukin-2Rα(-/-) mice[J]. J Autoimmun, 2014, 51: 99-108. DOI: 10.1016/j.jaut.2014.02.009. [16] LINDOR KD, GERSHWIN ME, POUPON R, et al. Primary biliary cirrhosis[J]. Hepatology (Baltimore, Md), 2009, 50(1): 291-308. DOI: 10.1002/hep.22906. [17] GAO CY, YAO Y, LI L, et al. Tissue-resident memory CD8+ T cells acting as mediators of salivary gland damage in a murine model of sjögren's syndrome[J]. Arthritis Rheumatol, 2019, 71(1): 121-132. DOI: 10.1002/art.40676. [18] SUN Y, ZHANG W, LI B, et al. The coexistence of Sjögren's syndrome and primary biliary cirrhosis: A comprehensive review[J]. Clin Rev Allergy Immunol, 2015, 48(2-3): 301-315. DOI: 10.1007/s12016-015-8471-1. [19] YAO Y, LI L, YANG SH, et al. CD8+ T cells and IFN-γ induce autoimmune myelofibrosis in mice[J]. J Autoimmun, 2018, 89: 101-111. DOI: 10.1016/j.jaut.2017.12.011. [20] ROMERO MF, FULTON CM, BORON WF. The SLC4 family of HCO3- transporters[J]. Pflugers Arch, 2004, 447(5): 495-509. DOI: 10.1007/s00424-003-1180-2. [21] GAWENIS LR, LEDOUSSAL C, JUDD LM, et al. Mice with a targeted disruption of the AE2 Cl-/HCO3- exchanger are achlorhydric[J]. J Biol Chem, 2004, 279(29): 30531-30539. DOI: 10.1074/jbc.M403779200. [22] BANALES JM, ARENAS F, RODRÍGUEZ-ORTIGOSA CM, et al. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger[J]. Hepatology, 2006, 43(2): 266-275. DOI: 10.1002/hep.21042. [23] SALAS JT, BANALES JM, SARVIDE S, et al. Ae2a, b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis[J]. Gastroenterology, 2008, 134(5): 1482-1493. DOI: 10.1053/j.gastro.2008.02.020. [24] RIEGER R, GERSHWIN ME. The X and why of xenobiotics in primary biliary cirrhosis[J]. J Autoimmun, 2007, 28(2-3): 76-84. DOI: 10.1016/j.jaut.2007.02.003. [25] RIEGER R, LEUNG PS, JEDDELOH MR, et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis[J]. J Autoimmun, 2006, 27(1): 7-16. DOI: 10.1016/j.jaut.2006.06.002. [26] AMANO K, LEUNG PS, RIEGER R, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: Identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid[J]. J Immunol, 2005, 174(9): 5874-5883. DOI: 10.4049/jimmunol.174.9.5874. [27] OPDYKE DL. Monographs on fragrance raw materials[J]. Food Cosmet Toxicol, 1979, 17(4): 357-390. DOI: 10.1016/0015-6264(79)90330-4. [28] WAKABAYASHI K, LIAN ZX, LEUNG PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease[J]. Hepatology, 2008, 48(2): 531-540. DOI: 10.1002/hep.22390. [29] WAKABAYASHI K, YOSHIDA K, LEUNG PS, et al. Induction of autoimmune cholangitis in non-obese diabetic (NOD). 1101 mice following a chemical xenobiotic immunization[J]. Clin Exp Immunol, 2009, 155(3): 577-586. DOI: 10.1111/j.1365-2249.2008.03837.x. [30] ALEXOPOULOU L, HOLT AC, MEDZHITOV R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3[J]. Nature, 2001, 413(6857): 732-738. DOI: 10.1038/35099560. [31] OKADA C, AKBAR SM, HORⅡKE N, et al. Early development of primary biliary cirrhosis in female C57BL/6 mice because of poly Ⅰ∶ C administration[J]. Liver Int, 2005, 25(3): 595-603. DOI: 10.1111/j.1478-3231.2005.01043.x. [32] JIANG T, HAN Z, CHEN S, et al. Resistance to activation-induced cell death and elevated FLIPL expression of CD4+ T cells in a polyI∶ C-induced primary biliary cirrhosis mouse model[J]. Clin Exp Med, 2009, 9(4): 269-276. DOI: 10.1007/s10238-009-0052-2. [33] AMBROSINI YM, YANG GX, ZHANG W, et al. The multi-hit hypothesis of primary biliary cirrhosis: Polyinosinic-polycytidylic acid (poly Ⅰ∶ C) and murine autoimmune cholangitis[J]. Clin Exp Immunol, 2011, 166(1): 110-120. DOI: 10.1111/j.1365-2249.2011.04453.x. [34] GERSHWIN ME, SELMI C, WORMAN HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients[J]. Hepatology, 2005, 42(5): 1194-1202. DOI: 10.1002/hep.20907. [35] HIRSCHFIELD GM, DYSON JK, ALEXANDER G, et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines[J]. Gut, 2018, 67(9): 1568-1594. DOI: 10.1136/gutjnl-2017-315259. -

 下载:
下载: