
 PDF下载 ( 2620 KB)
PDF下载 ( 2620 KB)

急性胰腺炎胰酶异常活化和分泌的分子机制
DOI: 10.3969/j.issn.1001-5256.2022.05.047
Research advances in abnormal activation and secretion of pancreatic enzymes in acute pancreatitis
-
摘要: 胰腺腺泡细胞内胰酶异常活化和分泌是急性胰腺炎(AP)的重要发病机制之一,可直接损伤胰腺组织加速疾病进程,诱发重症急性胰腺炎。目前临床抑制胰酶异常活化和分泌的药物效果欠佳,探寻新的治疗靶点十分重要。本文归纳了AP胰酶异常活化和分泌的病理事件(胞浆钙离子超载、溶酶体与酶原颗粒的共定位、细胞器损伤、胰酶顶端侧分泌受阻和基底侧分泌增加等),搜集了相关事件的分子机制,讨论了胰酶异常活化和分泌在AP早期的作用过程,为未来靶向药物的研发提供思路。Abstract: Abnormal activation and secretion of pancreatic enzymes in pancreatic acinar cells is one of the important pathogeneses of acute pancreatitis (AP) and can directly damage the pancreatic tissue to accelerate disease progression and induce severe AP. At present, the drugs inhibiting the abnormal activation and secretion of pancreatic enzymes tend to have an unsatisfactory effect in clinical practice, and therefore, it is of great importance to search for new therapeutic targets. This article summarizes the pathological events of abnormal activation and secretion of pancreatic enzymes (cytoplasmic calcium overload, colocalization of lysosomes and zymogen granules, organelle injury, obstructed apical secretion of trypsin, and increased basal secretion of trypsin), collects the molecular mechanisms of related events, and discusses the role of abnormal activation and secretion of pancreatic enzymes in the early stage of AP, so as to provide ideas for the development of targeted drugs in the future.
-
Key words:
- Pancreatitis /
- Pancreatin /
- Secretory Vesicles
-
急性胰腺炎(acute pancreatitis, AP)是一种临床常见急腹症,发病率为34/10万,在世界范围内呈上升趋势[1],其中重症急性胰腺炎(severe acute pancreatitis,SAP)常合并严重并发症,病死率达15%~35%[2-3],严重威胁人民生命健康,并带来巨大经济负担。这主要是由于缺乏治疗AP的特效药物,临床治疗仍以奥曲肽[4-5]、乌司他丁等药物治疗为主,但效果欠佳。因此深入探索AP发病机制,寻找理想的治疗靶点十分重要。
AP的发病机理十分复杂,其中胰酶异常活化和分泌是胰腺腺泡细胞中的早期病理事件[6],也是AP发生发展的始发因素。近年来,胰酶异常活化和分泌的细胞内分子机制研究取得新进展,本文将从胰腺腺泡细胞内病理事件和调控分子角度,归纳胰酶异常活化和分泌的机制。
1. AP胰酶异常活化的细胞内机制
胰腺外分泌部的主要生理功能是分泌胰液进入小肠,发挥消化吸收作用。胰蛋白酶原等胰酶前体是胰液的主要成分之一,由胰腺腺泡细胞合成、贮存和分泌。胰酶前体于腺泡细胞粗面内质网(endoplasmic reticulum,ER)中合成,经高尔基体加工后形成含有酶原颗粒(zymogen granules,ZG)的内吞囊泡。胰酶前体无活性,同时细胞内存在保护机制(活化后胰酶的自溶作用和特定胰酶抑制剂等)以避免胰酶前体激活,防止自身消化[7]损伤细胞。而AP早期病理损伤多由胰腺腺泡细胞中胰蛋白酶原提前激活引起。在AP相关病因(如胆结石和酒精滥用等)作用下,腺泡细胞内防止胰酶前体激活或降低胰酶活性的保护机制障碍,胰酶前体被提前活化为胰酶,进而多种酶(如弹性蛋白酶和磷脂酶A2)以及补体和激肽途径被激活,同时促炎因子分泌增加,造成胰腺局部损伤和炎症反应。目前胰酶异常活化的细胞机制主要包括钙离子(Ca2+)超载、溶酶体与ZG的共定位和细胞器损伤。
1.1 Ca2+超载
胰腺腺泡细胞内Ca2+信号调控是胰酶前体激活的关键信号分子,而Ca2+超载是AP发生的中心事件[8]。生理环境中,副交感神经末梢释放乙酰胆碱等刺激性神经递质,通过胰腺腺泡细胞膜上G蛋白偶联受体激活磷脂酶C,促进细胞膜表面的磷脂酰肌醇4,5二磷酸水解产生二级信使三磷酸肌醇(inositol 1,4,5-trisphosphate,InsP3)。InsP3作用于ER上三磷酸肌醇受体(inositol 1,4,5-trisphosphate receptors,InsP3Rs),打开InsP3依赖性钙通道,促进ER中大量Ca2+释放入细胞质中[9]; 同时细胞膜上Ca2+通道打开,胞外Ca2+内流,参与激活InsP3Rs和ryanodine受体,进一步促进Ca2+从ER释放。最终细胞外Ca2+内流和ER中Ca2+释放引起腺泡细胞顶端侧区域的Ca2+浓度瞬时峰值,启动酶原胞吐作用,并刺激线粒体中ATP的产生,为胰酶分泌提供能量[10]。并且胞外Ca2+内流和ER中Ca2+释放这两条路径之间存在相互作用。ryanodine受体是钙敏感通道,细胞内Ca2+浓度的轻度升高即可诱导其开放,介导Ca2+从ER释放。Orai1通道位于胞膜上,属于钙库调控的Ca2+通道,STIM1为Orai1通道的调控因子,位于ER膜上,可感知ER内Ca2+耗竭并向质膜迁移,与Orai1相互作用后打开钙库调控的Ca2+通道,诱导细胞外Ca2+内流[11]。
AP相关病因(高脂血症、酒精和胆汁酸等)刺激下,腺泡细胞内出现胞外Ca2+过量流入或胞质内Ca2+清除机制异常,造成胞浆内Ca2+超载。AP时细胞膜和ER膜的通透性增加,促进胞外和ER中Ca2+过量流入细胞质中。正常情况下,胞浆内Ca2+浓度一旦过度增加,细胞可通过ER和胞膜上的Ca2+泵迅速发挥代偿作用,分别将Ca2+移回ER或排出胞外[8],清除胞质内多余Ca2+,而Ca2+泵的代偿作用需要ATP供能维持。但是胞浆内Ca2+浓度持续性病理升高会导致线粒体通透性转换孔(mitochondrial permeability transition pore,MPTP)以高电导状态打开,损伤生成ATP[12-14]所需的膜电位,导致ATP耗竭,进而直接抑制腺泡细胞内Ca2+清除作用。Mukherjee等[12]也在CD1小鼠AP模型中证实胰腺腺泡细胞内Ca2+浓度持续升高可打开MPTP,造成胰腺腺泡细胞内线粒体损伤、ATP生产受阻; 敲除MPTP基因及药物阻滞MPTP后发现胰腺腺泡细胞内线粒体损伤减轻,ATP生成水平正常,进一步证实了抑制MPTP开放对AP线粒体损伤和ATP耗竭的保护作用。
AP时Ca2+超载可激活Ca2+/钙调蛋白依赖性丝氨酸/苏氨酸磷酸酶钙调神经磷酸酶(calmodulin-dependent serine/threonine phosphatase calcineurin,PP2B),促进酶原活化[7]。Shah等[15]利用PP2B抑制剂FK506分别于1 h和8 h干预AP小鼠,发现与1 h相比8 h小鼠胰腺内胰蛋白酶活性降低50%以上,血清淀粉酶降低86%,提示AP早期胰蛋白酶活化与PP2B活性相关。此外,Ca2+也可作为辅助因子参与胰酶自身激活过程,促进胰蛋白酶提前活化。虽然腺泡细胞内存在胰蛋白酶抑制剂,可通过降解胞质内低活性胰酶来自我保护,避免自消化损伤。但Ca2+可与胰蛋白酶激活肽的氮端天冬氨酸残基结合,增强胰酶的稳定性和活性,降低胰蛋白酶抑制剂对腺泡细胞的保护作用。已有研究[16]证实,增加的腺泡细胞内Ca2+的浓度可直接促进胰蛋白酶原的激活。Dantrolene是RyR的药理拮抗剂,Orabi等[17]使用Dantrolene(RyR拮抗剂)干预AP小鼠,通过减弱胰腺腺泡胞Ca2+内流降低胞浆内Ca2+浓度,发现酶原激活水平以及胰腺损伤程度均减轻。
1.2 溶酶体与ZG的共定位
胰酶以非活性ZG形式贮存于腺泡细胞胞质中。在AP早期阶段,腺泡细胞内溶酶体和ZG生成增加,并且二者发生融合现象,此过程称为共定位,导致胰蛋白酶原提前活化[6]。ZG与溶酶体融合后,组织蛋白酶B(CathepsinB, CatB)(一种主要的溶酶体酶)将胰蛋白酶原激活为具有消化功能的胰酶[18]。胰酶和CatB从内吞液泡内释放的机理尚未完全阐明,有研究者[19]认为活化后的胰酶可引起膜脆性的增加,导致内吞液泡泄漏,释放胰酶和CatB; 还有研究[20]认为,发生共定位的内吞液泡可能会诱使细胞骨架和/或细胞器破裂; 溶酶体和ZG膜也可能分别由于酒精的代谢产物和膜稳定糖蛋白2的缺失而变得脆弱。上述原因均可能促进内吞液泡释放胰酶,在腺泡细胞内外引起自身消化,造成胰腺损伤和全身炎症反应[21],加速AP的发生发展。
1.3 细胞器损伤
AP可通过损伤ER和线粒体等细胞器功能,促进胰酶提前激活。ER是调节蛋白质合成、折叠和聚集的重要细胞器。ER可通过泛素-蛋白酶体途径将未折叠或错误折叠的蛋白质清除,保证合成蛋白质的质量。外部刺激(如氧化应激或Ca2+超载)可引发ER中错误折叠的蛋白质过度聚集,导致内质网应激(endoplasmic reticulum stress,ERS)。ERS也是早期AP腺泡细胞中的重要反应,实验性AP中,早期ERS的发生与胰蛋白酶原激活有关[22-23]。AP腺泡细胞内线粒体损伤-ATP耗竭-胰酶激活过程与Ca2+超载密切相关。
ERS和线粒体损伤除直接参与胰酶激活外,也与AP自噬损伤有关。自噬是一种细胞保护机制,可处理和回收陈旧、有缺陷或已损坏的各种细胞质内容物[24]。AP腺泡细胞溶酶体功能障碍,促使溶酶体内组织蛋白酶L(降解胰蛋白酶原和胰酶,抑制CatB激活胰蛋白酶原)和CatB比例失衡,导致自噬损伤,促进自噬泡内胰蛋白酶原激活[25]。自噬受损也会加重ERS和线粒体功能损伤,导致腺泡细胞更易遭受其他损伤和死亡[26-29]。Hashimoto等[30]利用雨蛙素诱导自噬相关基因5敲除的AP小鼠模型发现,胰腺腺泡细胞内ZG的激活受到抑制,AP严重程度显著降低。
腺泡细胞中的细胞器形成了一个联动系统,任何细胞器受损可能都会导致整个网络的故障。例如,通过ATG5或ATG7的遗传消融或通过核因子κB激酶亚基抑制剂的基因缺失来阻止自噬,均导致ERS和线粒体功能紊乱,无法生成ATP,且由于胰蛋白酶原合成于ER,腺泡细胞合成和分泌胰酶功能也发生障碍[24, 31]。相反,通过CypD基因消融恢复线粒体功能可减轻ERS并增加实验性AP中溶酶体-自噬系统的性能[26]。虽然已有大量研究发现腺泡细胞细胞器之间的相互作用可影响胰酶活化,甚至影响AP病程发展,但其具体分子机制仍需深入挖掘。
2. AP胰酶分泌抑制的细胞机制
生理刺激下,胰腺腺泡细胞内顶端侧Ca2+浓度出现瞬时峰值,促使内吞囊泡向腺泡细胞顶端侧运输,最终与顶端侧胞膜融合,将酶原释放入腺泡管内(胰酶顶端侧分泌)。酶原后进入十二指肠,被肠激酶等消化酶活化后发挥生理功能。AP时患者胰酶肠道分泌减少,胰腺组织和血液中胰酶浓度增加,这是由腺泡细胞顶端侧分泌受阻以及基底侧分泌增强导致的。细胞内堆积的ZG被提前激活后进入胰腺组织,导致胰腺局部损伤和炎症反应; 进入血液后可造成全身炎症反应,促进SAP的发生发展。
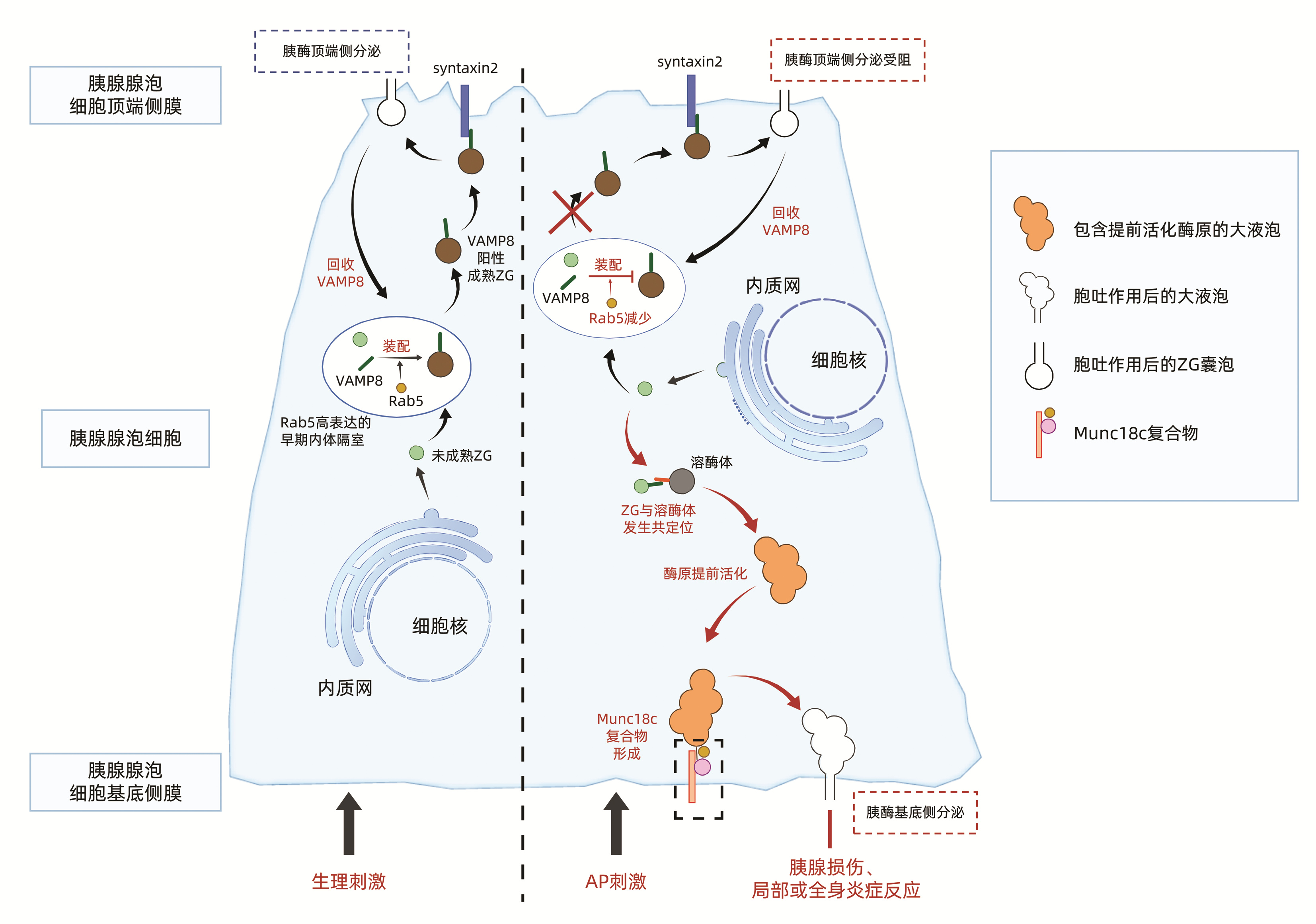
2.1 VAMP8介导的顶端侧分泌受损和基底侧分泌增强(图 1)
 图 1 VAMP8介导的顶端侧分泌受损和基底侧分泌增强示意图Figure 1. Schematic illustration of VAMP8-mediated impaired apical secretion and enhanced basal secretion
图 1 VAMP8介导的顶端侧分泌受损和基底侧分泌增强示意图Figure 1. Schematic illustration of VAMP8-mediated impaired apical secretion and enhanced basal secretionVAMP(vesicle associated membrane protein,VAMP)8和VAMP2属于SNARE家族,广泛存在于包裹ZG的囊泡(内体)膜上,VAMP8和VAMP2是正常生理状态下介导胰酶顶端侧分泌的主要蛋白。然而AP时VAMP8介导的腺泡细胞胰酶顶端侧分泌路径完全阻断,仅50%胰酶通过VAMP2介导的顶端侧分泌,导致细胞内ZG堆积[32],胰酶分泌受阻。堆积的ZG更易与溶酶体发生共定位,导致胰酶提前活化。Messenger等[32]证实,超大剂量胰酶分泌激动剂刺激野生型和VAMP8基因敲除小鼠后,VAMP8基因敲除AP小鼠分离的胰腺腺泡细胞内ZG堆积水平较野生型小鼠增加了4.5倍,但ZG与溶酶体共定位减少,最终腺泡细胞内胰酶活性无明显变化,提示VAMP8除参与ZG顶端侧分泌外,还可能与囊泡间融合相关。
正常胰腺腺泡细胞中ZG分泌后,顶端侧扩大的早期内体隔室通过Rab5介导的内体顺行运输,回收质膜和VAMP8等胞吐相关蛋白,并且Rab5介导的内体顺行运输系统可促进ZG装配VAMP8,维持VAMP8介导的ZG顶端侧分泌。说明在生理状态下胰腺腺泡细胞VAMP8介导的ZG顶端侧分泌依赖于Rab5参与调控的内体运输[33]。AP时Rab5减少,导致VAMP8介导的ZG顶端侧分泌路径受损,ZG转而与晚期内体、溶酶体融合并提前活化,向基底侧逆向转运[34]。Messenger等[34-35]还发现无胰酶分泌激动剂刺激时,Rab5过表达能促进VAMP8介导的ZG顶端侧分泌,上调胰酶基础分泌; Rab5过表达也可减少ZG与溶酶体融合,抑制胰酶提前活化。
2.2 肌动蛋白细胞骨架破坏和融合孔扩张不足
肌动蛋白丝在调节胰腺腺泡细胞顶端侧分泌中起重要作用,超最大剂量的雨蛙素可造成末端肌动蛋白网及其相关的中间丝的损失。Chvanov等[36]于免疫电镜下观察发现,过度刺激下胰腺腺泡细胞内肌动蛋白丝无法完整包裹ZG运载囊泡,进而囊泡出现相互融合、破裂,最终ZG顶端侧分泌胞吐作用受损; 然而基底侧胞膜肌动蛋白丝增加,这可能与胰酶基底侧增强有关。因此胰酶分泌阻滞可能是与胆囊收缩素过量刺激破坏肌蛋白细胞骨架有关,但详细分子调控尚不明确。
生理状态下,运载ZG的内吞囊泡与腺泡细胞顶端膜结合后形成足够大的融合孔,酶原通过融合孔释放入腺泡管内,目前认为依赖ATP供能的融合孔扩张在ZG分泌中起关键作用。AP致使融合孔扩张不足,造成酶原等大分子蛋白质无法通过融合孔,顶端侧分泌受阻,也有研究者认为融合孔扩张不足与细胞内Ca2+超载引起的ATP耗竭有关[37],融合孔扩张不足的具体机制同样不明确。
3. 结语
胰酶异常活化和分泌是AP发生发展的关键因素。早期胰腺腺泡细胞内Ca2+超载、溶酶体与ZG的共定位和细胞器损伤均可导致胰酶提前活化; 而胰酶顶端侧分泌受阻和基底侧分泌增加、肌动蛋白细胞骨架破坏和融合孔扩张不足可致胰酶异常分泌。胰酶异常活化和分泌可直接促进全身炎症反应和多器官系统损伤的发生,导致SAP,增加病死率。因此深入探索胰酶异常活化和分泌的细胞机制,寻找早期阻断AP发生发展的靶向治疗药物十分关键,可有效降低SAP的发生率和病死率。
-
[1] LEE PJ, PAPACHRISTOU GI. New insights into acute pancreatitis[J]. Nat Rev Gastroenterol Hepatol, 2019, 16(8): 479-496. DOI: 10.1038/s41575-019-0158-2. [2] ZHANG W, ZHANG M, KUANG Z, et al. The risk factors for acute respiratory distress syndrome in patients with severe acute pancreatitis: A retrospective analysis[J]. Medicine (Baltimore), 2021, 100(2): e23982. DOI: 10.1097/MD.0000000000023982. [3] LEPPÄNIEMI A, TOLONEN M, TARASCONI A, et al. 2019 WSES guidelines for the management of severe acute pancreatitis[J]. World J Emerg Surg, 2019, 14: 27. DOI: 10.1186/s13017-019-0247-0. [4] HAO S, DANG DS. Clinical observation of octreotide in the treatment of acute pancreatitis at different time of administration[J]. Chin J Med Offic, 2020, 48(7): 761-764. DOI: 10.16680/j.1671-3826.2020.07.04.郝双, 党大胜. 奥曲肽不同给药时机治疗急性胰腺炎临床效果观察[J]. 临床军医杂志, 2020, 48(7): 761-764. DOI: 10.16680/j.1671-3826.2020.07.04. [5] QI H, CHEN G, WANG YW, et al. Effects of magnesium isoglycyrrhizinate combined with octreotide on acute pancreatitis and its effect on hs-CRP、IL-1β、IL-6 levels[J]. J Changchun Univ Chin Med, 2020, 36(3): 520-522. DOI: 10.13463/j.cnki.cczyy.2020.03.032.齐宏, 陈广, 王渊文, 等. 异甘草酸镁联合奥曲肽治疗急性胰腺炎的临床疗效及对hs-CRP、IL-1β、IL-6水平的影响[J]. 长春中医药大学学报, 2020, 36(3): 520-522. DOI: 10.13463/j.cnki.cczyy.2020.03.032. [6] GUKOVSKAYA AS, GORELICK FS, GROBLEWSKI GE, et al. Recent insights into the pathogenic mechanism of pancreatitis: Role of acinar cell organelle disorders[J]. Pancreas, 2019, 48(4): 459-470. DOI: 10.1097/MPA.0000000000001298. [7] MAYERLE J, SENDLER M, HEGYI E, et al. Genetics, cell biology, and pathophysiology of pancreatitis[J]. Gastroenterology, 2019, 156(7): 1951-1968. e1. DOI: 10.1053/j.gastro.2018.11.081. [8] PALLAGI P, MADÁCSY T, VARGA Á, et al. Intracellular Ca2+ signalling in the pathogenesis of acute pancreatitis: Recent advances and translational perspectives[J]. Int J Mol Sci, 2020, 21(11): 4005. DOI: 10.3390/ijms21114005. [9] WENG N, BAUMLER MD, THOMAS DD, et al. Functional role of J domain of cysteine string protein in Ca2+-dependent secretion from acinar cells[J]. Am J Physiol Gastrointest Liver Physiol, 2009, 296(5): G1030-1039. DOI: 10.1152/ajpgi.90592.2008. [10] CRIDDLE DN, MCLAUGHLIN E, MURPHY JA, et al. The pancreas misled: Signals to pancreatitis[J]. Pancreatology, 2007, 7(5-6): 436-446. DOI: 10.1159/000108960. [11] LEE KP, YUAN JP, HONG JH, et al. An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs[J]. FEBS Lett, 2010, 584(10): 2022-2027. DOI: 10.1016/j.febslet.2009.11.078. [12] MUKHERJEE R, MARENINOVA OA, ODINOKOVA IV, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: Inhibition prevents acute pancreatitis by protecting production of ATP[J]. Gut, 2016, 65(8): 1333-1346. DOI: 10.1136/gutjnl-2014-308553. [13] PETERSEN OH, GERASIMENKO JV, GERASIMENKO OV, et al. The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas[J]. Physiol Rev, 2021, 101(4): 1691-1744. DOI: 10.1152/physrev.00003.2021. [14] ELMUNZER BJ, SERRANO J, CHAK A, et al. Rectal indomethacin alone versus indomethacin and prophylactic pancreatic stent placement for preventing pancreatitis after ERCP: Study protocol for a randomized controlled trial[J]. Trials, 2016, 17(1): 120. DOI: 10.1186/s13063-016-1251-2. [15] SHAH AU, SARWAR A, ORABI AI, et al. Protease activation during in vivo pancreatitis is dependent on calcineurin activation[J]. Am J Physiol Gastrointest Liver Physiol, 2009, 297(5): G967-973. DOI: 10.1152/ajpgi.00181.2009. [16] MITHÖFER K, FERNÁNDEZ-DEL CASTILLO C, FRICK TW, et al. Acute hypercalcemia causes acute pancreatitis and ectopic trypsinogen activation in the rat[J]. Gastroenterology, 1995, 109(1): 239-246. DOI: 10.1016/0016-5085(95)90290-2. [17] ORABI AI, SHAH AU, AHMAD MU, et al. Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice[J]. Am J Physiol Gastrointest Liver Physiol, 2010, 299(1): G196-204. DOI: 10.1152/ajpgi.00498.2009. [18] HALANGK W, LERCH MM, BRANDT-NEDELEV B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis[J]. J Clin Invest, 2000, 106(6): 773-781. DOI: 10.1172/JCI9411. [19] TALUKDAR R, SAREEN A, ZHU H, et al. Release of cathepsin B in cytosol causes cell death in acute pancreatitis[J]. Gastroenterology, 2016, 151(4): 747-758. e5. DOI: 10.1053/j.gastro.2016.06.042. [20] SALUJA A, DUDEJA V, DAWRA R, et al. Early intra-acinar events in pathogenesis of pancreatitis[J]. Gastroenterology, 2019, 156(7): 1979-1993. DOI: 10.1053/j.gastro.2019.01.268. [21] SENDLER M, WEISS FU, GOLCHERT J, et al. Cathepsin B-mediated activation of trypsinogen in endocytosing macrophages increases severity of pancreatitis in mice[J]. Gastroenterology, 2018, 154(3): 704-718. e10. DOI: 10.1053/j.gastro.2017.10.018. [22] CHENG L, HAN ST. Research Progress on the mechanism of endoplasmic reticulum stress response in acute pancreatitis[J]. Chin J Pancreatol, 2019, 19(1): 69-72. DOI: 10.3760/cma.j.issn.1674-1935.2019.01.018.程璐, 韩树堂. 急性胰腺炎内质网应激反应机制研究进展[J]. 中华胰腺病杂志, 2019, 19(1): 69-72. DOI: 10.3760/cma.j.issn.1674-1935.2019.01.018. [23] SZMOLA R, SAHIN-TÓTH M. Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells[J]. Gut, 2010, 59(3): 365-372. DOI: 10.1136/gut.2009.198903. [24] ANTONUCCI L, FAGMAN JB, KIM JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress[J]. Proc Natl Acad Sci U S A, 2015, 112(45): E6166-6174. DOI: 10.1073/pnas.1519384112. [25] WARTMANN T, MAYERLE J, KÄHNE T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice[J]. Gastroenterology, 2010, 138(2): 726-737. DOI: 10.1053/j.gastro.2009.10.048. [26] BICZO G, VEGH ET, SHALBUEVA N, et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models[J]. Gastroenterology, 2018, 154(3): 689-703. DOI: 10.1053/j.gastro.2017.10.012. [27] DENG SJ, ZOU WB, LIAO Z. Research progress on the mechanism of carboxyl ester lipase in the pathogenesis of pancreatic diseases[J]. Chin J Pancreatol, 2020, 20(1): 66-69. DOI: 10.3760/cma.j.issn.1674-1935.2020.01.014.邓顺江, 邹文斌, 廖专. 羧基酯脂肪酶在胰腺疾病发病中的作用机制研究进展[J]. 中华胰腺病杂志, 2020, 20(1): 66-69. DOI: 10.3760/cma.j.issn.1674-1935.2020.01.014. [28] LUGEA A, GERLOFF A, SU HY, et al. The combination of alcohol and cigarette smoke induces endoplasmic reticulum stress and cell death in pancreatic acinar cells[J]. Gastroenterology, 2017, 153(6): 1674-1686. DOI: 10.1053/j.gastro.2017.08.036. [29] ZELIC M, RODERICK JE, O'DONNELL JA, et al. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome[J]. J Clin Invest, 2018, 128(5): 2064-2075. DOI: 10.1172/JCI96147. [30] HASHIMOTO D, OHMURAYA M, HIROTA M, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells[J]. J Cell Biol, 2008, 181(7): 1065-1072. DOI: 10.1083/jcb.200712156. [31] LI N, WU X, HOLZER RG, et al. Loss of acinar cell IKKα triggers spontaneous pancreatitis in mice[J]. J Clin Invest, 2013, 123(5): 2231-2243. DOI: 10.1172/JCI64498. [32] MESSENGER SW, JONES EK, HOLTHAUS CL, et al. Acute acinar pancreatitis blocks vesicle-associated membrane protein 8 (VAMP8)-dependent secretion, resulting in intracellular trypsin accumulation[J]. J Biol Chem, 2017, 292(19): 7828-7839. DOI: 10.1074/jbc.M117.781815. [33] MIZUNO-YAMASAKI E, RIVERA-MOLINA F, NOVICK P. GTPase networks in membrane traffic[J]. Annu Rev Biochem, 2012, 81: 637-659. DOI: 10.1146/annurev-biochem-052810-093700. [34] MESSENGER SW, FALKOWSKI MA, THOMAS DD, et al. Vesicle associated membrane protein 8 (VAMP8)-mediated zymogen granule exocytosis is dependent on endosomal trafficking via the constitutive-like secretory pathway[J]. J Biol Chem, 2014, 289(40): 28040-28053. DOI: 10.1074/jbc.M114.593913. [35] MESSENGER SW, THOMAS DD, COOLEY MM, et al. Early to late endosome trafficking controls secretion and zymogen activation in rodent and human pancreatic acinar cells[J]. Cell Mol Gastroenterol Hepatol, 2015, 1(6): 695-709. DOI: 10.1016/j.jcmgh.2015.08.002. [36] CHVANOV M, DE FAVERI F, MOORE D, et al. Intracellular rupture, exocytosis and actin interaction of endocytic vacuoles in pancreatic acinar cells: Initiating events in acute pancreatitis[J]. J Physiol, 2018, 596(13): 2547-2564. DOI: 10.1113/JP275879. [37] NEULAND K, FRICK M. Vesicular control of fusion pore expansion[J]. Commun Integr Biol, 2015, 8(2): e1018496. DOI: 10.1080/19420889.2015.1018496. 期刊类型引用(9)
1. 莫闲,杨闯. 急性胰腺炎发病机制及并发内脏静脉血栓研究进展. 陕西医学杂志. 2024(02): 282-285 .  百度学术
百度学术2. 杨晶晶,高翔. 腺泡细胞钙离子超载与急性胰腺炎关系的研究进展. 中国当代医药. 2024(01): 192-198 . 百度学术3. 孙敏慧,沈红璋,张筱凤. 胰管支架置入在预测为重症急性胆源性胰腺炎中的疗效分析. 中华消化内镜杂志. 2024(03): 218-223 . 百度学术4. 谭文君,邢斌瑜,王赛宇,李豹,陈霖玉,王文静. 大承气汤联合穴位贴敷治疗急性胰腺炎. 吉林中医药. 2024(09): 1060-1063 . 百度学术5. 彭爽,孙泽林,廖蝶,杨晓淋,柯路. CRAC通道在胰腺炎发病机制中作用的研究进展. 中国病理生理杂志. 2024(12): 2351-2358+2366 . 百度学术6. 刘波,刘力华. 急性胰腺炎患者血清STAT3、HMGB1水平及其临床意义. 检验医学与临床. 2023(21): 3109-3113 . 百度学术7. 张琦,田朝霞,佟明铭,李坤,陶红. 胰炎消方辅助治疗急性胰腺炎的疗效及对sFlt-1、α-MSH的影响. 湖南中医药大学学报. 2023(11): 2048-2053 . 百度学术8. 刘静,罗宇维,郑彦涛,刘斌,王烁,赵义博,王永东. 肝素结合蛋白在急性胰腺炎合并感染中的预测价值评估的Meta分析. 现代疾病预防控制. 2023(11): 841-845+874 . 百度学术9. 杨波,王朝英,雷婷,熊元武,蔡小军. LDH、HMGB-1联合AChE活性检测在急性胰腺炎病情、预后评估中的应用. 解放军医药杂志. 2022(06): 48-51 . 百度学术其他类型引用(4)
-

 下载:
下载:

 下载:
下载:
 百度学术
百度学术

