| [1] |
|
| [2] |
MEMON N, WEINBERGER BI, HEGYI T, et al. Inherited disorders of bilirubin clearance[J]. Pediatr Res, 2016, 79(3): 378-386. DOI: 10.1038/pr.2015.247. |
| [3] |
STRASSBURG CP. Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome)[J]. Best Pract Res Clin Gastroenterol, 2010, 24(5): 555-571. DOI: 10.1016/j.bpg.2010.07.007. |
| [4] |
MARUO Y, BEHNAM M, IKUSHIRO S, et al. Two different UGT1A1 mutations causing Crigler-Najjar syndrome types I and Ⅱ in an Iranian family[J]. J Gastrointestin Liver Dis, 2015, 24(4): 523-526. DOI: 10.15403/jgld.2014.1121.244.ugt. |
| [5] |
BAI J, ZHENG SJ, DUAN ZP. Clinical features and diagnosis of four common types of congenital hyperbilirubinemia[J]. J Clin Hepatol, 2019, 35(8): 1680-1683. DOI: 10.3969/j.issn.1001-5256.2019.08.005. |
| [6] |
DHAWAN A, LAWLOR MW, MAZARIEGOS GV, et al. Disease burden of Crigler-Najjar syndrome: Systematic review and future perspectives[J]. J Gastroenterol Hepatol, 2020, 35(4): 530-543. DOI: 10.1111/jgh.14853. |
| [7] |
KRINGEN MK, PIEHLER AP, GRIMHOLT RM, et al. Serum bilirubin concentration in healthy adult North-Europeans is strictly controlled by the UGT1A1 TA-repeat variants[J]. PLoS One, 2014, 9(2): e90248. DOI: 10.1371/journal.pone.0090248. |
| [8] |
ERLINGER S, ARIAS IM, DHUMEAUX D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences[J]. Gastroenterology, 2014, 146(7): 1625-1638. DOI: 10.1053/j.gastro.2014.03.047. |
| [9] |
CHRISTOFORIDOU A, FASOULAKIS Z, KONTOMANOLIS EN. Diagnosis and management of congenital dyserythropoietic anaemia Type Ⅱ in a secundigravida[J]. Cureus, 2017, 9(10): e1811. DOI: 10.7759/cureus.1811. |
| [10] |
BERGMANN C, GUAY-WOODFORD LM, HARRIS PC, et al. Polycystic kidney disease[J]. Nat Rev Dis Primers, 2018, 4(1): 50. DOI: 10.1038/s41572-018-0047-y. |
| [11] |
BOTSTEIN D, RISCH N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease[J]. Nat Genet, 2003, 33 Suppl: 228-237. DOI: 10.1038/ng1090. |



 本站查看
本站查看
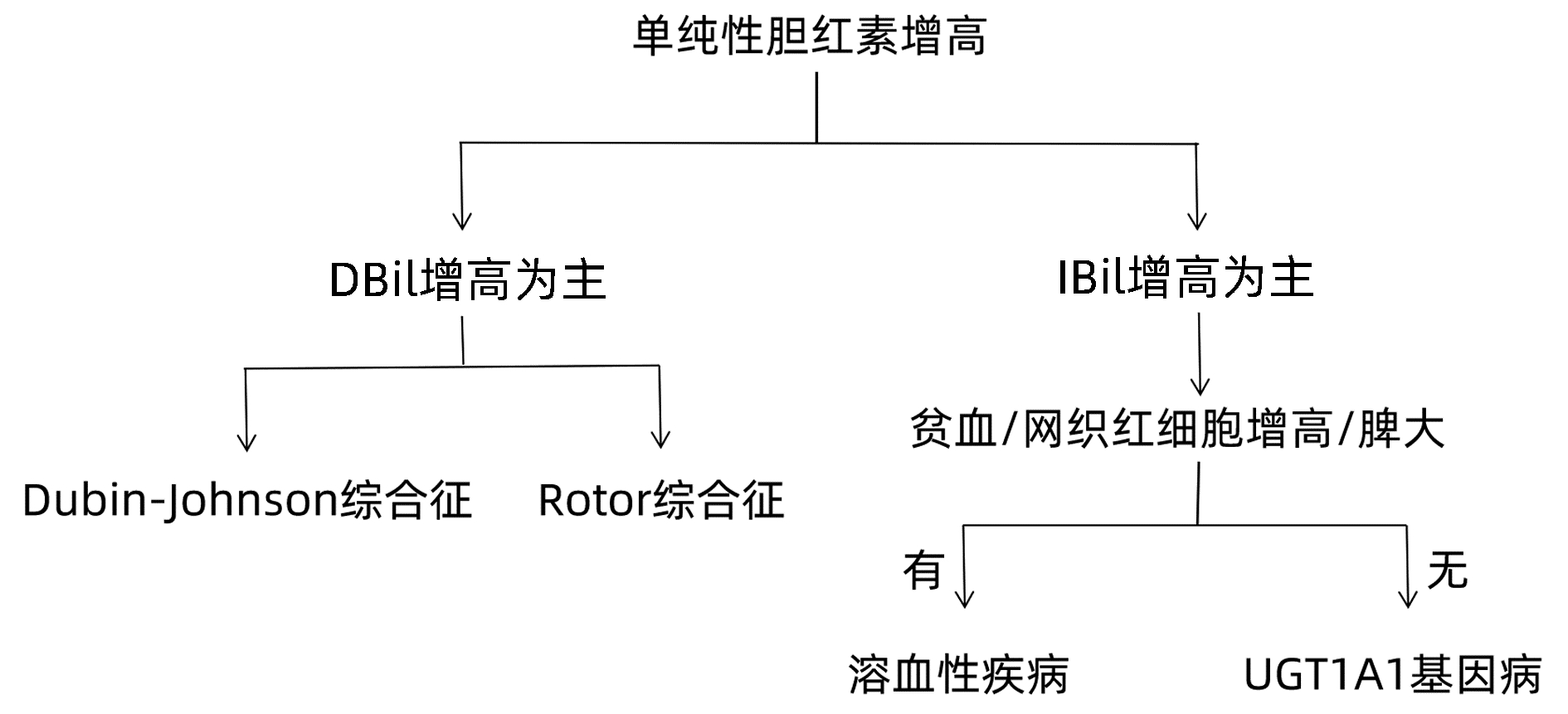




 DownLoad:
DownLoad: