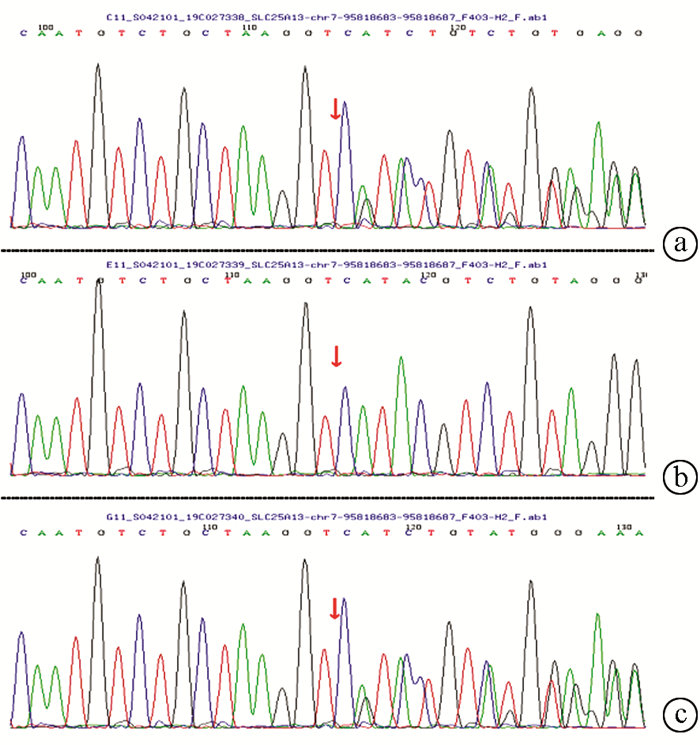
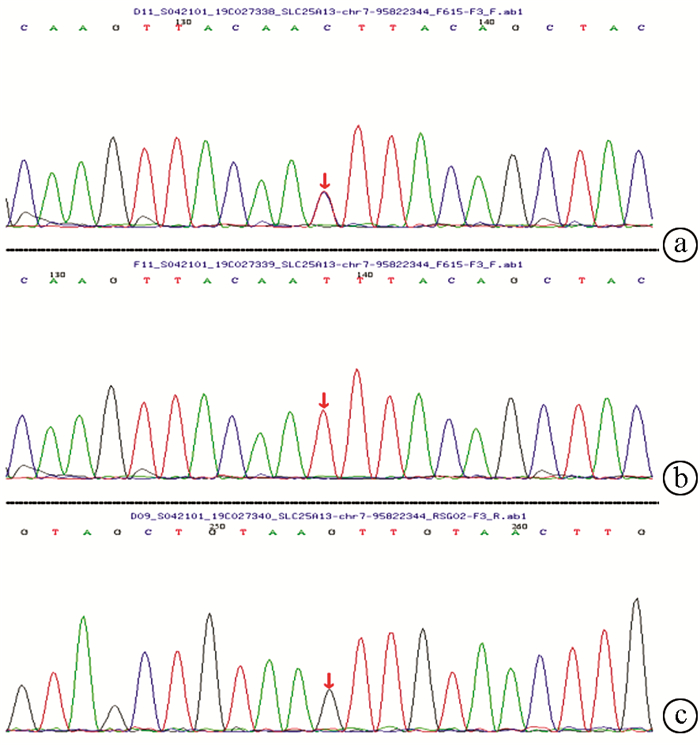
| [1] |
KOBAYASHI K, SINASAC DS, ⅡJIMA M, et al. The gene mutated in adult-onset type Ⅱ citrullinaemia encodes a putative mitochondrial carrier protein[J]. Nat Genet, 1999, 22(2): 159-163. DOI: 10.1038/9667. |
| [2] |
SONG YZ, DENG M, CHEN FP, et al. Genotypic and phenotypic features of citrin deficiency: Five year experience in a Chinese pediatric center[J]. Int J Mol Med, 2011, 28(1): 3340. DOI: 10.3892/ijmm.2011.653. |
| [3] |
SONG YZ, LI BX, HAO H, et al. Selective screening for inborn errors of metabolism and secondary methylmalonic aciduria in pregnancy at high risk district of neural tube defects: A human metabolome study by GC-MS in China[J]. Clin Biochem, 2008, 41(7-8): 616-620. DOI: 10.1016/j.clinbiochem.2008.01.025. |
| [4] |
KOBAYASHI K, ⅡJIMA M, USHIKAI M, et al. Citrin deficiency[J]. J Jpn Pediatr Soc, 2006, 110: 1047-1059.
|
| [5] |
KIM Y, CHOI JY, LEE SH, et al. Malfunction in mitochondrial β-oxidation contributes to lipid accumulation in hepatocyte-like cells derived from citrin deficiency-induced pluripotent stem cells[J]. Stem Cells Dev, 2016, 25(8): 636-647. DOI: 10.1089/scd.2015.0342. |
| [6] |
TANG CF, LIU SC, FENG Y, et al. Newborn screening program and blood amino acid profiling in early neonates with citrin deficiency[J]. Chin J Pediatr, 2019, 57(10): 797-801. DOI: 10.3760/cma.j.issn.0578-1310.2019.10.014. |
| [7] |
CHEW HB, NGU LH, ZABEDAH MY, et al. Neonatal intrahepatic cholestasis associated with citrin deficiency (NICCD): A case series of 11 Malaysian patients[J]. J Inherit Metab Dis, 2010, 33(Suppl 3): S489-S495. DOI: 10.1007/s10545-010-9248-6. |
| [8] |
SHIGEMATSU Y, HIRANO S, HATA I, et al. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2002, 776(1): 39-48. DOI: 10.1016/s1570-0232(02)00077-6. |
| [9] |
|
| [10] |
ZHANG ZH, LIN WX, ZHENG QQ, et al. Molecular diagnosis of citrin deficiency in an infant with intrahepatic cholestasis: Identification of a 21.7kb gross deletion that completely silences the transcriptional and translational expression of the affected SLC25A13 allele[J]. Oncotarget, 2017, 8(50): 87182-87193. DOI: 10.18632/oncotarget.19901. |
| [11] |
SONG YZ, ZHANG ZH, LIN WX, et al. SLC25A13 gene analysis in citrin deficiency: Sixteen novel mutations in East Asian patients, and the mutation distribution in a large pediatric cohort in China[J]. PLoS One, 2013, 8(9): e74544. DOI: 10.1371/journal.pone.0074544. |
| [12] |
LIN WX, ZENG HS, ZHANG ZH, et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution[J]. Sci Rep, 2016, 6: 29732. DOI: 10.1038/srep29732. |
| [13] |
LIN WX, ZENG HS, ZHANG ZH, et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution[J]. Sci Rep, 2016, 6: 29732. DOI: 10.1038/srep29732. |
| [14] |
WANG L, CHENG XR, YAN L, et al. Analysis of clinical features and SLC25A13 gene mutations in a family affected with neonatal intrahepatic cholestasis[J]. Chin J Med Genetics, 2016, 33(5): 670-673. DOI: 10.3760/cma.j.issn.1003-9406.2016.05.020. |
| [15] |
ZHANG JL, SHU SN, CAI ZS, et al. Neonatal intrahepatic cholestasis caused by Citrin deficiency with hepatic cirrhosis ascites as the main manifestation: A case report[J]. J Clin Hepatol, 2019, 35(2): 372-375. DOI: 10.3969/j.issn.1001-5256.2019.02.026. |



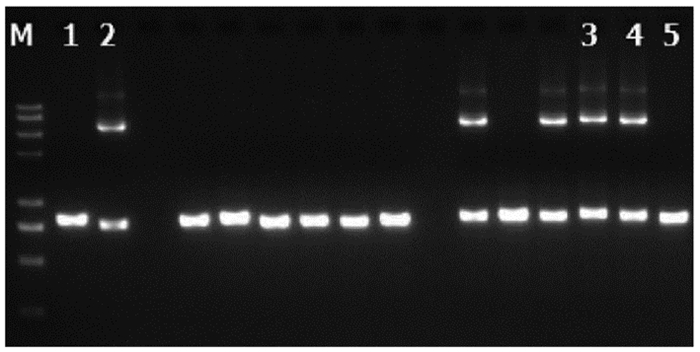




 DownLoad:
DownLoad: